
-
摘要:
癌细胞是一类分裂增殖能力强大的恶性细胞,癌细胞依靠大量消耗人体营养来维持自身分裂增殖所需的能量。癌细胞的分裂增殖离不开细胞核内遗传物质的合成与复制,阻断或者破坏癌细胞遗传物质的合成是大部分抗肿瘤药物的作用机制之一。细胞核遗传物质作为主导细胞分裂、增殖、死亡等过程的关键物质,对细胞最终的命运走向具有决定性作用。细胞核遗传物质主要是指位于细胞核中染色质上的脱氧核糖核酸,癌细胞遗传物质损伤后其最终的命运走向如何,值得研究和分析。因此本文选择细胞周期阻滞、细胞凋亡、细胞自噬、细胞衰老等方面对癌细胞遗传物质损伤后细胞最终的命运走向进行粗略论述及分析,以期为抗肿瘤药物的机制研究提供思路。
Abstract:Cancer cells refer to a group of malignant cells with strong division and proliferation abilities. Cancer cells rely on the unstable plunder of human nutrition to sustain the large amount of energy that they need for their own division and proliferation. The division and proliferation of cancer cells are linked to the synthesis and replication of genetic material in the nucleus. Blockage or destruction of the synthesis of genetic material in cancer cells is one of the mechanisms underlying the action of most antitumor drugs. As the key material that dominates cell division, proliferation, and death, nuclear genetic material which mainly refers to the deoxyribonucleic acid located on the chromatin in the nucleus, plays a decisive role in the final fate of cells. The final fate of cancer cells after the damage of the genetic material is worthy of investigation and analysis. In this paper, we discuss and analyze the fate of cancer cells after genetic material damage from the aspect of cellular cycle arrest, apoptosis, autophagy, and senescence to provide ideas for the mechanism research on antitumor drugs.
-
Key words:
- Genetic material damage /
- Cancer cell /
- Cycle arrest /
- Apoptosis /
- Autophagy /
- Senescence
-
0 引言
依据现代分子生物学理论,癌症基本上来说是一种基因疾病,是身体中某些细胞在癌基因的驱动下发生不受机体控制的恶性增殖,且人体免疫系统又无法杀灭此种恶性增殖细胞,最后大量消耗机体能量,影响相应脏器的功能,造成持续的炎性反应,最后导致人体组织器官破坏、免疫系统崩塌、能量耗竭的一种疾病[1]。根据遗传学定律,承载癌细胞遗传物质的脱氧核糖核酸(deoxyribo-nucleic acid, DNA)是癌细胞持续分裂产生更多子代癌细胞的关键,而癌细胞DNA链因为缺少管家基因的约束又时刻处于不停的突变和基因修复中[2]。针对癌细胞这一特性,研发了多种抗癌药物用于干预癌细胞的基因组,如与癌细胞DNA发生共价结合的烷化剂、干扰癌细胞DNA合成的抗代谢类抗肿瘤药物等[3]。那么细胞核遗传物质受到损伤后的癌细胞最终命运如何?癌细胞是通过自我损伤修复,最后从生存危机中幸存下来继续生长,还是囿于遗传物质损伤严重最后在危机中死亡?这很可能取决于遗传物质的修复情况是否可以满足癌细胞正常分裂所需要的最低要求[4]。下文将通过细胞周期阻滞、细胞凋亡、细胞自噬、细胞衰老等方面分析癌细胞在细胞核遗传物质损伤后的不同命运归属。
1 细胞核遗传物质损伤与细胞周期阻滞
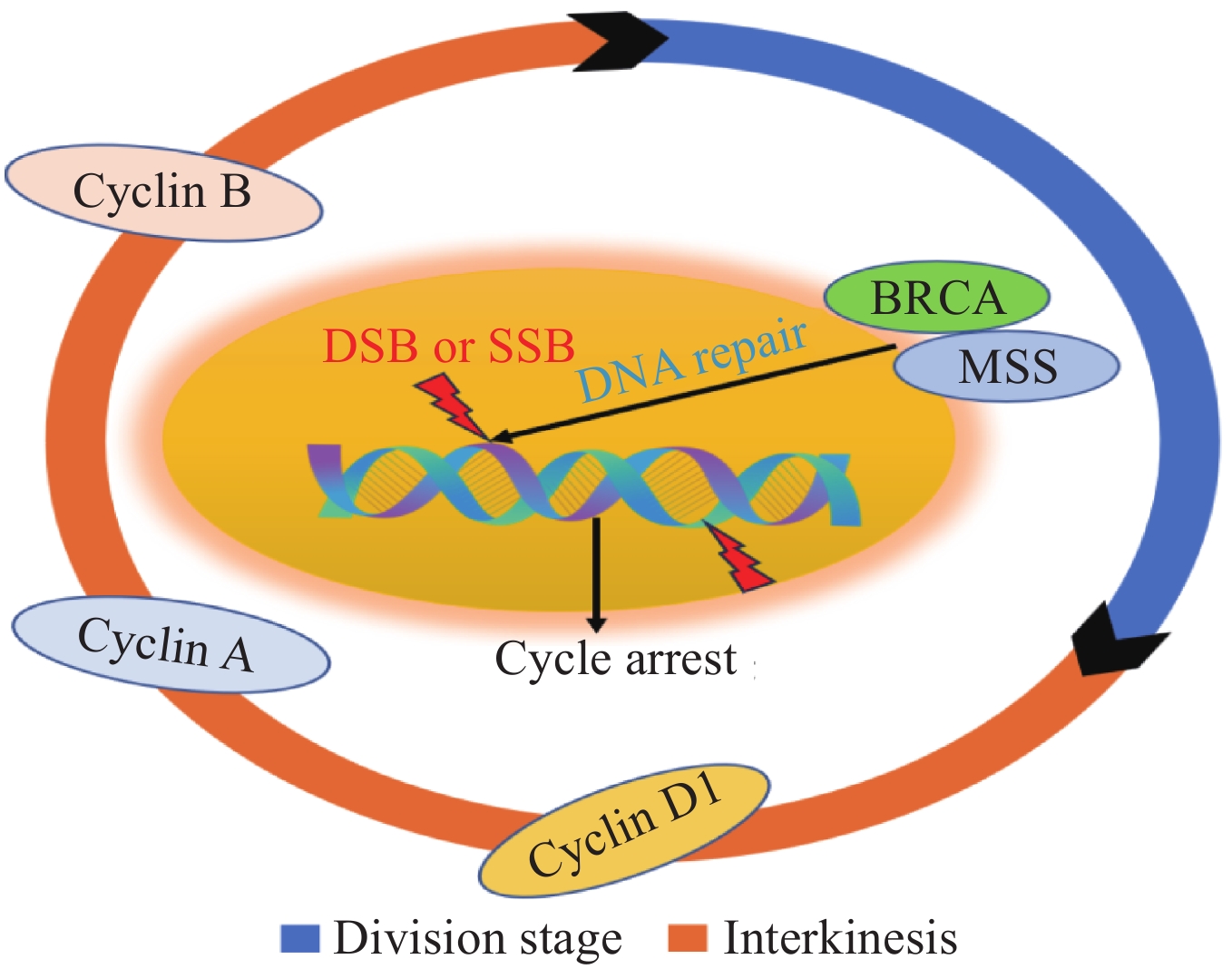
细胞分裂周期是指细胞从一次有丝分裂完成开始到下一次有丝分裂结束所经历的全过程,可分为分裂间期和分裂期。分裂间期又可以分为G1期、S期、G2期三个周期时相,分别受到不同的细胞周期激酶(cyclin-dependent kinase, CDK)调控,G1/S期由细胞周期蛋白D1(cyclin D1)和细胞周期蛋白E(cyclin E)调控,S/G2期由细胞周期蛋白A(cyclin A)调控,G2/M期由细胞周期蛋白B(cyclin B)调控[5-6]。细胞分裂间期占据了细胞分裂周期的绝大部分时间,细胞分裂间期主要完成DNA的合成与复制、相关蛋白质的翻译,并为下一步分裂期做好充足的准备。癌细胞分裂增殖明显较正常细胞更快,其细胞分裂增殖周期更短,很多抗肿瘤药物对癌细胞的抑制作用,可能都具有阻碍癌细胞分裂增殖、拉长癌细胞分裂周期的作用[7]。根据抗癌药物作用于肿瘤细胞的周期时相可以分为:细胞周期非特异性抗癌药和细胞周期特异性抗癌药,前者对于癌细胞增殖周期的各个时相或静止期细胞均有杀灭作用,后者仅针对癌细胞增殖周期的特定时相[8]。
当癌细胞核遗传物质受到物理、化学或者生物性损伤后,其DNA链出现断裂,包括DNA单链断裂(single-strand breakage, SSB)和DNA双链断裂(double-strand breakage, DSB),一般情况下DSB的修复难度明显大于SSB[9]。由于DNA链断裂,此时处于分裂周期的细胞因模板DNA链损伤,无法根据碱基互补配对原则完成子代DNA链的复制,则细胞分裂间期时间被拉长,出现细胞周期阻滞[10]。癌细胞因DNA受损无法进入分裂期,细胞被阻滞于分裂间期,此时细胞自身会通过多种途径和方式来对受损的遗传物质进行修复,包括DNA的切除修复、DNA重组修复、逆转录等[11-12]。同时癌细胞在完成自身修复的过程中募集了大量的遗传物质修复酶促进损伤修复[13],若某些损伤修复酶的基因出现突变(如BRCA基因突变、微卫星高度不稳定),则细胞损伤修复难度陡增,细胞在持续的细胞周期阻滞中出现严重的生存危机,见图1。可以说细胞周期阻滞是所有癌细胞细胞核遗传物质受损后必定会出现的过程,对遗传物质修复的情况决定了癌细胞命运的最终走向[14]。当癌细胞因为细胞核遗传物质损伤后出现持续的细胞周期阻滞,可以说此时癌细胞已经来到了生死存亡的关键路口,细胞自身能够完成损伤修复,则进入正常的细胞周期,完成下一轮的细胞增殖,反之则进入细胞的程序性死亡等过程,最后诱导癌细胞崩解发生细胞死亡。针对癌细胞周期的诸多抗癌药物早已被发现和深入研究,迄今仍然被广泛用于抗肿瘤的临床治疗,为肿瘤患者的临床治疗发挥重要作用。
2 细胞核遗传物质损伤与细胞凋亡
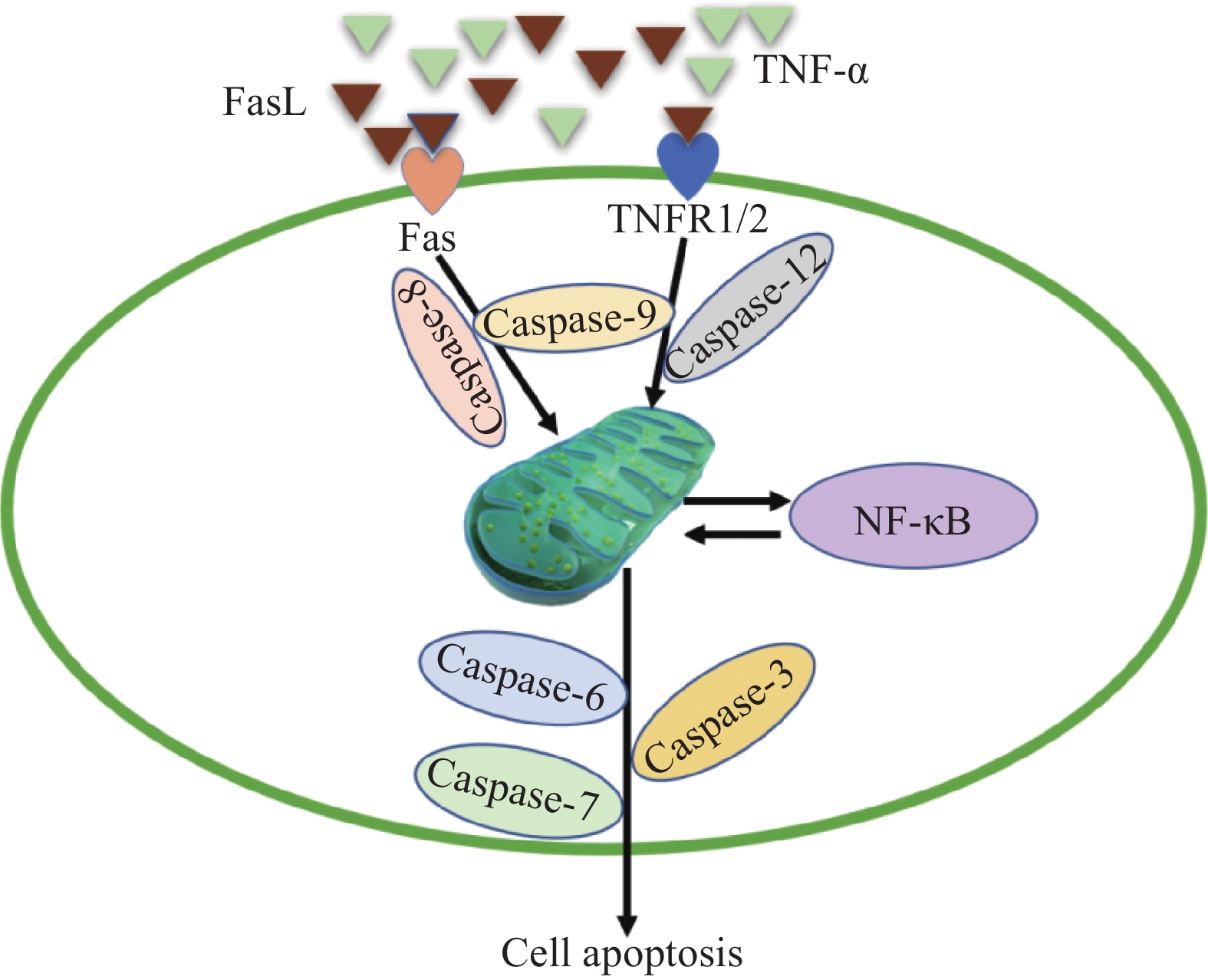
细胞凋亡(cellular apoptosis)是经典的细胞程序性死亡途径之一,细胞凋亡过程中涉及一系列的基因激活、表达等过程,细胞在凋亡相关蛋白酶的作用和调控下出现主动的死亡过程[15]。我们所熟知的凋亡途径主要是在三磷酸腺苷(adenosine triphosphate, ATP)供能下完成的,称为线粒体凋亡途径,细胞接受到凋亡信号,如肿瘤坏死因子受体(tumornecrosis factor receptor,TNFR)等后,诱导细胞激活凋亡相关信号通路,继而活化蛋白水解酶,最后进入细胞程序性死亡的级联反应,诱导细胞核固缩、破裂细胞结构、裂解核纤维、DNA片段化等[16]。细胞凋亡的核心调控因子是含半胱氨酸的天冬氨酸蛋白水解酶(cysteinyl aspartate specific proteinase, Caspase)家族,包括与上游促进凋亡信号联系密切的“启动类”Caspase如Caspase-8/9/12等[17-18],以及下游的“执行类”Caspase如Caspase-3/6/7等[19]。上游的“启动类”Caspase通过接受外源性死亡受体介导细胞程序性死亡途径,外源性死亡受体主要由TNFR家族成员(TNFR1/2、Fas)及相关配体(TNF-α、FasL)构成[20]。同时经典的凋亡途径受到核因子-κB(nuclear factor kappa B,NF-κB)的调节,在某些情况下NF-κB可以使处于凋亡程序中的细胞幸存下来,也可以协助凋亡途径促进癌细胞死亡,具有双向调节作用[21],见图2。
细胞核遗传物质损伤与细胞凋亡关系密切,核内遗传物质是细胞分裂增殖最重要的结构,当癌细胞细胞核遗传物质受损后,亲代细胞无法完成DNA的合成与复制,导致子代DNA链合成障碍[22]。如果细胞核遗传物质在癌细胞激发的自我修复过程中仍然无法完成修复,不能够满足DNA复制的要求,则癌细胞仍处于持续的细胞周期停滞中,此时部分癌细胞在接收到凋亡信号后,为了癌细胞种群的增殖,选择性牺牲遗传物质受损严重的癌细胞,诱发癌细胞的程序性死亡[23]。特别是某些特定基因分型的癌细胞(如BRCA基因突变的卵巢癌细胞、微卫星高度不稳定的结直肠癌细胞)很难对受损的细胞核遗传物质进行修复,这类细胞对遗传物质损伤特别敏感,遗传物质受损后也极容易诱发细胞凋亡[24]。但值得注意的是,并非细胞核遗传物质受损后,细胞一定会出现程序性死亡途径,很多时候癌细胞会借助自身和人体正常细胞参与细胞核遗传物质的损伤修复,以帮助癌细胞度过危机[25]。也有可能癌细胞会选择性舍弃某段受损遗传物质,在某些酶的协助下进行遗传物质重组,化解遗传物质受损带来的生存危机[26]。度过生存危机(凋亡失败)后的癌细胞的恶性程度及侵袭能力很有可能得到强化[27]。可以说细胞凋亡是癌细胞为了更好地适应环境、利于种群更好地增殖而主动争取的一种死亡过程。但是如何诱导肿瘤细胞发生更多的程序性死亡仍然是临床肿瘤治疗的关键,通过药物或其他治疗方式持续性对肿瘤细胞遗传物质进行破坏,从而诱导癌细胞发生凋亡也是抗肿瘤治疗最常见的手段及方式。
3 细胞核遗传物质损伤与细胞自噬
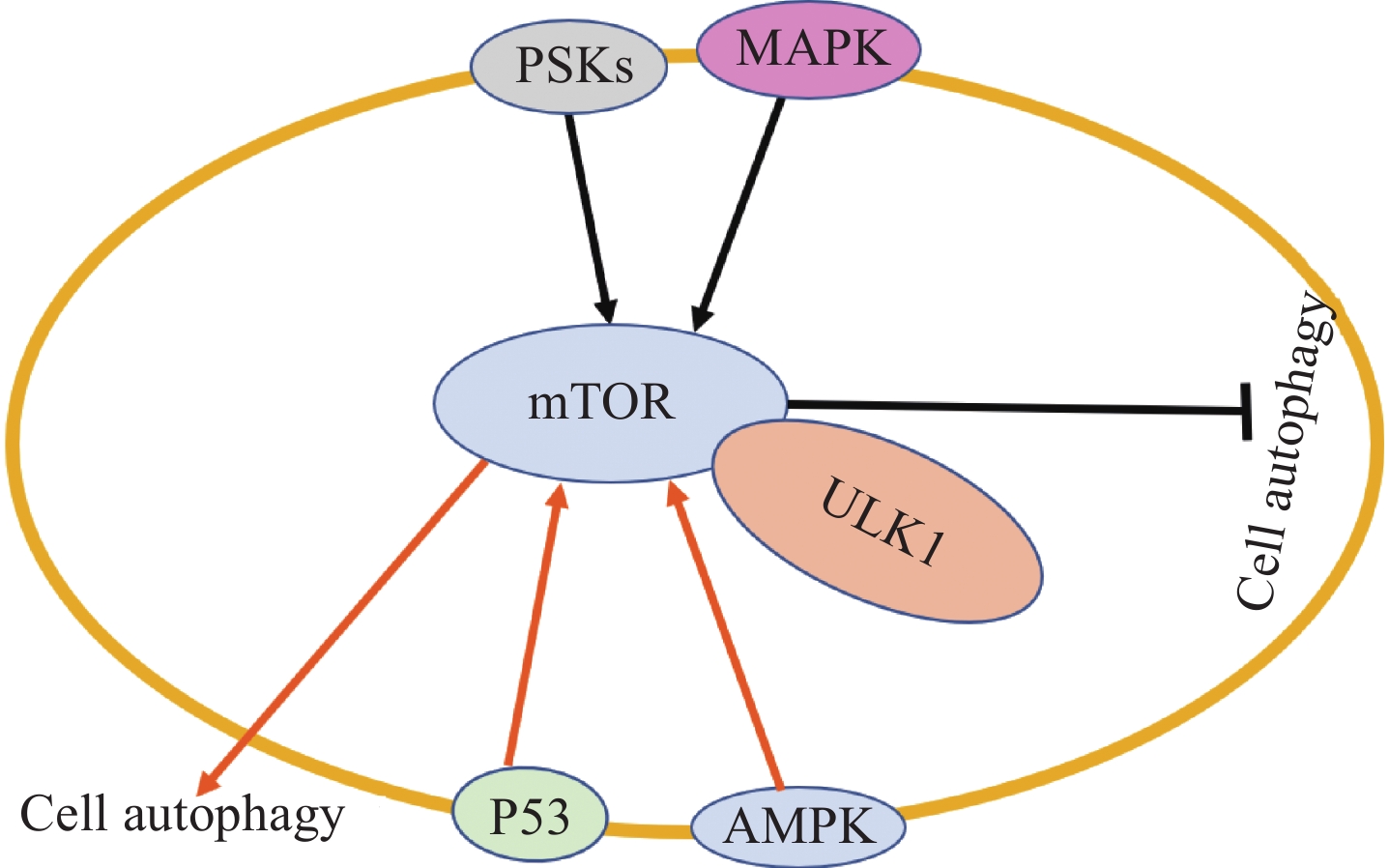
自噬(cellular autophagy)是另外一种细胞程序性死亡途径,是细胞在多种压力条件下启动的一种生理过程,主要过程包括降解或者消除受损的细胞器、错误的DNA产物,促使细胞度过当前的艰难环境,达到存活的目的[28]。自噬过程中自噬体将降解的细胞内物质运输到溶酶体,以便细胞在不利生存条件下实现物质循环利用,这种机制可以极大程度保护细胞免受毒素攻击、维持细胞能量和代谢稳定,这是促进细胞存活至关重要的过程[29]。细胞通过自噬途径可以清除细胞内受到损伤和衰老的成分,起到更新细胞结构的作用[30]。同时细胞可通过自噬途径降解细胞内非必要成分,给自身提供生存所必须的物质达到“自给自足”状态,帮助细胞度过生存危机[31]。细胞自噬途径中哺乳动物雷帕霉素靶蛋白(mammalian target of rapamycin, mTOR)通路是最为关键的节点之一,激活mTOR通路的丝裂原活化蛋白激酶(mitogen-activated protein kinase, MAPK)和丝氨酸/苏氨酸激酶(protein serine/threonine kinases, PSKs)信号抑制细胞自噬,抑制mTOR通路的磷酸腺苷依赖蛋白激酶(AMP-activated protein kinase, AMPK)和P53信号促进细胞自噬[32]。在危机条件刺激下细胞内mTOR失活,此时活化的AMPK催化自噬相关蛋白抗体(Unc-51-like Kinase 1, ULK1)相应位点的丝氨酸发生磷酸化而促进自噬,见图3。当细胞危机条件解除后,AMPK失活mTOR与ULK1相应位点丝氨酸结合抑制ULK1-AMPK相互作用,关闭自噬信号通路[33]。
细胞核遗传物质损伤作为癌细胞处于生存危机的诱发因素之一,与细胞自噬关系密切[34]。当癌细胞在某些压力条件作用下细胞核遗传物质受到损伤,细胞DNA复制及蛋白质合成障碍无法满足正常的分裂增殖需要,癌细胞如何度过此时的生存危机,细胞自噬很好地回答了这一个问题。细胞自噬是癌细胞应对威胁性应激源的重要途径之一,自噬与癌细胞对抗肿瘤药的耐药性形成关系密切,且近年来发现自噬可能参与了癌细胞的免疫逃逸[35]。加拿大玛格丽特公主癌症中心的科学家Catherine O'Brien博士近期在Cell和Nature上发文指出:当受到威胁时,所有癌细胞都有能力进入一种“保护状态”,在这种状态下癌细胞可进入一种可逆的周期停滞,癌细胞借此“蒙混过关”躲过免疫系统的识别,威胁解除后癌细胞可被唤醒进入分裂旺盛期,并很可能在此过程中获得对抗肿瘤药的耐药性[36-37]。总的来说细胞自噬为癌细胞在遗传物质受损后的命运走向上提供了另外一种可能的路径,当然也为理解癌细胞生物学特性提供了更深的认识,同时也为临床治疗癌症提供了新的机会[38]。如何抑制癌细胞遗传物质损伤后的自噬路径,是最大化临床抗肿瘤治疗的可行方向之一,类似的治疗方案可以在抗肿瘤治疗的路径上加载抑制自噬的药物,这可以最大化诱导肿瘤细胞死亡。可以预见靶向癌细胞自噬途径的药物将会在未来进入广泛的研究,为恶性肿瘤的治愈带来更多的可能。
4 细胞核遗传物质损伤与细胞衰老
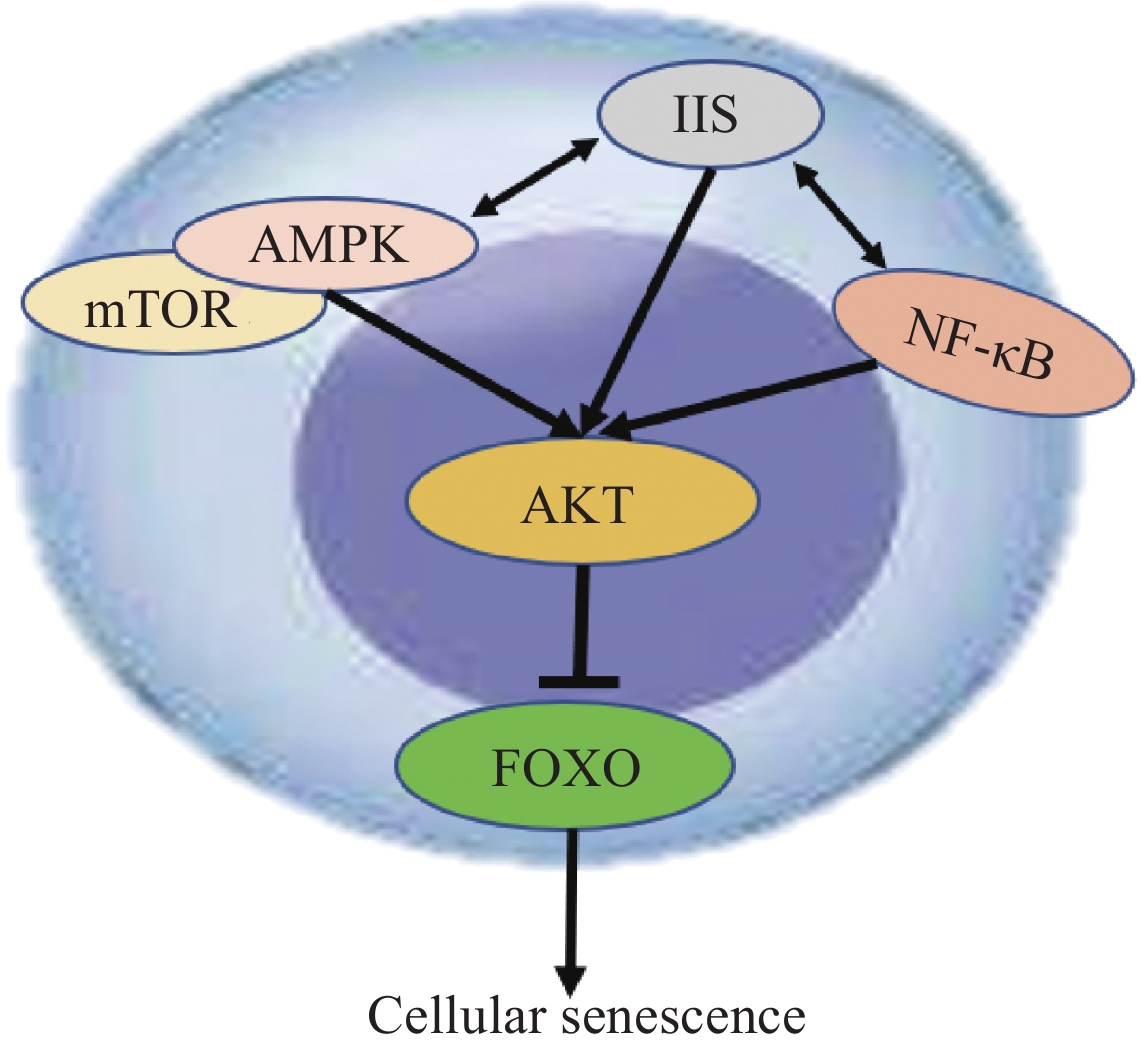
细胞衰老是细胞在生命活动过程中随着时间推移以及外界环境影响,出现增殖分化停滞、细胞生理功能衰退、细胞衰老死亡的一系列生命过程[39]。细胞衰老过程中可见细胞核、细胞质、细胞膜等细胞器的明显变化,包括染色质固缩、碎裂;细胞质色素沉积、空泡形成;线粒体减少;高尔基体消失;细胞膜粘度增加等细胞形态变化[40]。细胞衰老在分子水平上可表现为蛋白质合成减少、结构稳定性降低,DNA氧化、断裂、缺失、交联等,关键活性酶被氧化、二级结构改变等异常[41]。细胞衰老过程中涉及多条经典信号通路及转录因子的调控,目前认为参与细胞衰老途径的信号通路包括胰岛素/胰岛素样生长因子1信号通路(insulin/IGF-1 signaling,IIS)、AMPK-mTOR信号通路、核因子-κB信号通路(nuclear factor kappa B,NF-κB)等[42]。IIS信号通路是最早定义与衰老和年龄相关的信号转导途径,由胰岛素/胰岛素样肽与细胞表面相应受体结合后触发细胞内酶级联反应,最后活化丝氨酸/苏氨酸激酶(RAC-alpha serine/threonine-protein kinase,AKT)抑制叉头转录因子(forkhead box O,FOXO),促进细胞衰老进程[43]。IIS信号通路还可与AMPK-mTOR和NF-κB等信号通路交联形成调节细胞寿命和衰老的复杂网络[44],见图4。
细胞核遗传物质损伤可诱导癌细胞进入细胞衰老途径,细胞核遗传物质作为细胞分裂增殖的关键,癌细胞遗传物质受到损伤后细胞处于严重危机之中,细胞衰老可作为癌细胞暂时度过生存危机的途径之一[45]。细胞衰老可导致癌细胞处于细胞周期停滞状态(也称细胞永久性周期阻滞),此时癌细胞对促进生长的信号产生耐受,导致衰老细胞代谢减弱、染色质重构、基因表达改变,并伴随多种促进细胞炎性反应表型的发生[46]。诱导细胞衰老发生是治疗肿瘤的又一方法,当前的研究认为诱导癌细胞衰老并选择性杀死衰老癌细胞可作为一种抗肿瘤的有效方法[47]。但是应该引起重视的是细胞衰老有可能促进上皮间质转化(epithelial-mesenchymal transition, EMT)导致肿瘤转移风险升高,并且可募集某些细胞因子来促进血管和淋巴管形成,为肿瘤的生长或转移提供帮助[48]。总的来说细胞衰老作为癌细胞遗传物质损伤后可能启动的应对途径之一具有重要的研究价值。诚然,简单地认为癌细胞衰老可以抵抗肿瘤的生长是比较肤浅的,癌细胞衰老所诱发的其他复杂反应对整个肿瘤细胞群体的作用也应该引起重视。但遗憾的是,迄今为止,可以靶向细胞衰老的抗肿瘤药物仍然相当匮乏,导致抗肿瘤治疗效果相对弱化,如何针对癌细胞这一途径开发相应的治疗方法是优化临床抗肿瘤治疗的方向之一。
5 小结与展望
癌细胞因为强大的分裂增殖能力,其遗传物质的合成与复制均较正常细胞明显加快,造成癌细胞遗传物质损伤是抗肿瘤治疗最常见的作用途径。临床上很多抗肿瘤药物均对癌细胞遗传物质具有损伤作用,理论上来说癌细胞遗传物质受到损伤后其增殖分裂会受到明显控制,但遗憾的是临床上更多见到的是肿瘤患者对抗肿瘤治疗后的耐受,最后出现肿瘤进展。如何理解癌细胞在细胞核遗传物质损伤后的命运走向,成为了认识这种残酷临床现象的关键。本文通过细胞周期阻滞、细胞凋亡、细胞自噬、细胞衰老等四个方面粗略分析了癌细胞在细胞核遗传物质损伤后的命运走向。
我们认为细胞周期阻滞是癌细胞在细胞核遗传物质损伤后最常见的细胞响应,而细胞衰老则是永久性的细胞周期阻滞;细胞凋亡和细胞自噬则是癌细胞为了度过生存危机、保障自身种群生长所主动选择的细胞程序性途径,在凋亡和自噬途径后幸存的癌细胞很大可能会获得对该种生存危机的耐受。也就是说在癌细胞未获得抗性之前,细胞周期阻滞和细胞衰老可以明显抑制肿瘤分裂增殖,这也是临床上肿瘤患者最初接受抗肿瘤治疗比较敏感的原因。而随着抗肿瘤治疗的推进,肿瘤患者体内的癌细胞在凋亡和自噬途径中少部分幸存了下来,获得了对当前治疗的耐受性,则肿瘤患者逐渐出现对当前治疗的不敏感,直至最后肿瘤进展。更加遗憾的是周期阻滞、凋亡、自噬、衰老等并不能完全解释癌细胞遗传物质损伤后所表现出来的全部响应,似乎癌细胞仍然存在着其他应对遗传物质损伤的响应机制。
癌细胞是在人体正常细胞的基础上恶性转化而来的,对我们人体高度熟悉,它们已经演化出来了各种应对抗肿瘤治疗的生存策略,加大对癌细胞生物学特性的研究仍然是治疗恶性肿瘤最关键的前提条件。虽然目前对癌细胞生物学行为和特征的研究已经有所突破,但仍然无法从根本上治愈分期较晚的恶性肿瘤,如何治愈晚期癌症仍然任重道远。相信随着对癌细胞生物学行为和特性的深入研究,揭示癌细胞更多不为人知的特性和行为,临床上治愈恶性肿瘤的愿景是可以触及的。
Competing interests: The authors declare that they have no competing interests.利益冲突声明:所有作者均声明不存在利益冲突。作者贡献:王 磊:提出思路、稿件撰写许小敏:文献查阅、稿件审核王建、牟方政、魏大荣:论文指导 -
[1] Pavlova NN, Zhu J, Thompson CB. The hallmarks of cancer metabolism: Still emerging[J]. Cell Metab, 2022, 34(3): 355-377. doi: 10.1016/j.cmet.2022.01.007
[2] Hanahan D. Hallmarks of Cancer: New Dimensions[J]. Cancer Discov, 2022, 12(1): 31-46. doi: 10.1158/2159-8290.CD-21-1059
[3] Huang X, Ke K, Jin W, et al. Identification of Genes Related to 5-Fluorouracil Based Chemotherapy for Colorectal Cancer[J]. Front Immunol, 2022, 13: 887048. doi: 10.3389/fimmu.2022.887048
[4] Ryu JY, Oh J, Kim SM, et al. SOCS1 counteracts ROS-mediated survival signals and promotes apoptosis by modulating cell cycle to increase radiosensitivity of colorectal cancer cells[J]. BMB Rep, 2022, 55(4): 198-203. doi: 10.5483/BMBRep.2022.55.4.191
[5] Jun SY, Kim J, Yoon N, et al. Prognostic Potential of Cyclin D1 Expression in Colorectal Cancer[J]. J Clin Med, 2023, 12(2): 572. doi: 10.3390/jcm12020572
[6] Yan H, Jiang F, Yang J. Association of β-Catenin, APC, SMAD3/4, Tp53, and Cyclin D1 Genes in Colorectal Cancer: A Systematic Review and Meta-Analysis[J]. Genet Res (Camb), 2022, 2022: 5338956.
[7] Catanzaro D, Milani G, Bozza A, et al. Selective cell cycle arrest in glioblastoma cell lines by quantum molecular resonance alone or in combination with temozolomide[J]. Br J Cancer, 2022, 127(5): 824-835. doi: 10.1038/s41416-022-01865-9
[8] Sun J, Li M, Lin T, et al. Cell cycle arrest is an important mechanism of action of compound Kushen injection in the prevention of colorectal cancer[J]. Sci Rep, 2022, 12(1): 4384. doi: 10.1038/s41598-022-08336-4
[9] Swift ML, Azizkhan-Clifford J. DNA damage-induced sumoylation of Sp1 induces its interaction with RNF4 and degradation in S phase to remove 53BP1 from DSBs and permit HR[J]. DNA Repair (Amst), 2022, 111: 103289. doi: 10.1016/j.dnarep.2022.103289
[10] Dullovi A, Ozgencil M, Rajvee V, et al. Microtubule-associated proteins MAP7 and MAP7D1 promote DNA double-strand break repair in the G1 cell cycle phase[J]. iScience, 2023, 26(3): 106107. doi: 10.1016/j.isci.2023.106107
[11] Gupta D, Beisel CL. Illuminating the path to DNA repair[J]. Cell, 2021, 184(22): 5503-5505. doi: 10.1016/j.cell.2021.10.005
[12] Desai RV, Chen X, Martin B, et al. A DNA repair pathway can regulate transcriptional noise to promote cell fate transitions[J]. Science, 2021, 373(6557): eabc6506. doi: 10.1126/science.abc6506
[13] Lu C, Guan J, Lu S, et al. DNA Sensing in Mismatch Repair-Deficient Tumor Cells Is Essential for Anti-tumor Immunity[J]. Cancer Cell, 2021, 39(1): 96-108. e6.
[14] Wang D, Wu W, Callen E, et al. Active DNA demethylation promotes cell fate specification and the DNA damage response[J]. Science, 2022, 378(6623): 983-989. doi: 10.1126/science.add9838
[15] Liao Q, Ren Y, Yang Y, et al. CCT8 recovers WTp53-suppressed cell cycle evolution and EMT to promote colorectal cancer progression[J]. Oncogenesis, 2021, 10(12): 84. doi: 10.1038/s41389-021-00374-3
[16] Ripani P, Delp J, Bode K, et al. Thiazolides promote G1 cell cycle arrest in colorectal cancer cells by targeting the mitochondrial respiratory chain[J]. Oncogene, 2020, 39(11): 2345-2357. doi: 10.1038/s41388-019-1142-6
[17] Kim SJ, Kang CH, Kim GH, et al. Anti-Tumor Effects of Heat-Killed L. reuteri MG5346 and L. casei MG4584 against Human Colorectal Carcinoma through Caspase-9-Dependent Apoptosis in Xenograft Model[J]. Microorganisms, 2022, 10(3): 533. doi: 10.3390/microorganisms10030533
[18] Piao MJ, Han X, Kang KA, et al. The Endoplasmic Reticulum Stress Response Mediates Shikonin-Induced Apoptosis of 5-Fluorouracil-Resistant Colorectal Cancer Cells[J]. Biomol Ther (Seoul), 2022, 30(3): 265-273. doi: 10.4062/biomolther.2021.118
[19] Nasrullah U, Stanke K, Recknagel V, et al. The E3 Ligase TRIM25 Impairs Apoptotic Cell Death in Colon Carcinoma Cells via Destabilization of Caspase-7 mRNA: A Possible Role of hnRNPH1[J]. Cells, 2023, 12(1): 201. doi: 10.3390/cells12010201
[20] Bakshi HA, Quinn GA, Nasef MM, et al. Crocin Inhibits Angiogenesis and Metastasis in Colon Cancer via TNF-α/NF-kB/VEGF Pathways[J]. Cells, 2022, 11(9): 1502. doi: 10.3390/cells11091502
[21] Papierska K, Krajka-Kuźniak V, Kleszcz R, et al. The synthesis of novel thioderivative chalcones and their influence on NF-κB, STAT3 and NRF2 signaling pathways in colorectal cancer cells[J]. Sci Rep, 2022, 12(1): 14915. doi: 10.1038/s41598-022-18981-4
[22] Lu H, Yang M, Zhou Q. Reprogramming transcription after DNA damage: recognition, response, repair, and restart[J]. Trends Cell Biol, 2022, 33(8): 682-694.
[23] Etourneaud L, Moussa A, Rass E, et al. Lamin B1 sequesters 53BP1 to control its recruitment to DNA damage[J]. Sci Adv, 2021, 7(35): eabb3799. doi: 10.1126/sciadv.abb3799
[24] Scott DE, Francis-Newton NJ, Marsh ME, et al. A small-molecule inhibitor of the BRCA2-RAD51 interaction modulates RAD51 assembly and potentiates DNA damage-induced cell death[J]. Cell Chem Biol, 2021, 28(6): 835-847. e5.
[25] Liu L, Dai X, Yin S, et al. DNA-PK promotes activation of the survival kinase AKT in response to DNA damage through an mTORC2-ECT2 pathway[J]. Sci Signal, 2022, 15(715): eabh2290. doi: 10.1126/scisignal.abh2290
[26] Luan Y, Yu SY, Abazarikia A, et al. TAp63 determines the fate of oocytes against DNA damage[J]. Sci Adv, 2022, 8(51): eade1846. doi: 10.1126/sciadv.ade1846
[27] Berthenet K, Castillo Ferrer C, Fanfone D, et al. Failed Apoptosis Enhances Melanoma Cancer Cell Aggressiveness[J]. Cell Rep, 2020, 31(10): 107731. doi: 10.1016/j.celrep.2020.107731
[28] Kataura T, Sedlackova L, Otten EG, et al. Autophagy promotes cell survival by maintaining NAD levels[J]. Dev Cell, 2022, 57(22): 2584-2598. doi: 10.1016/j.devcel.2022.10.008
[29] Deretic V. Autophagy in inflammation, infection, and immunometabolism[J]. Immunity, 2021, 54(3): 437-453. doi: 10.1016/j.immuni.2021.01.018
[30] Levine B, Kroemer G. Biological Functions of Autophagy Genes: A Disease Perspective[J]. Cell, 2019, 176(1-2): 11-42. doi: 10.1016/j.cell.2018.09.048
[31] Galluzzi L, Green DR. Autophagy-Independent Functions of the Autophagy Machinery[J]. Cell, 2019, 177(7): 1682-1699. doi: 10.1016/j.cell.2019.05.026
[32] Pei F, Ma L, Jing J, et al. Sensory nerve niche regulates mesenchymal stem cell homeostasis via FGF/mTOR/autophagy axis[J]. Nat Commun, 2023, 14(1): 344. doi: 10.1038/s41467-023-35977-4
[33] Karabiyik C, Vicinanza M, Son SM, et al. Glucose starvation induces autophagy via ULK1-mediated activation of PIKfyve in an AMPK-dependent manner[J]. Dev Cell, 2021, 56(13): 1961-1975. e5.
[34] Wang W, Li J, Tan J, et al. Endonuclease G promotes autophagy by suppressing mTOR signaling and activating the DNA damage response[J]. Nat Commun, 2021, 12(1): 476. doi: 10.1038/s41467-020-20780-2
[35] Yamamoto K, Venida A, Yano J, et al. Autophagy promotes immune evasion of pancreatic cancer by degrading MHC-I[J]. Nature, 2020, 581(7806): 100-105. doi: 10.1038/s41586-020-2229-5
[36] Rehman SK, Haynes J, O'Brien CA, et al. Colorectal Cancer Cells Enter a Diapause-like DTP State to Survive Chemotherapy[J]. Cell, 2021, 184(1): 226-242. e21.
[37] Rehman SK, O'Brien CA. Persister cells that survive chemotherapy are pinpointed[J]. Nature, 2022, 608(7924): 675-676.
[38] Xia H, Green DR, Zou W. Autophagy in tumour immunity and therapy[J]. Nat Rev Cancer, 2021, 21(5): 281-297. doi: 10.1038/s41568-021-00344-2
[39] Aghali A, KolokoNgassie ML, Pabelick CM, et al. Cellular Senescence in Aging Lungs and Diseases[J]. Cells, 2022, 11(11): 1781. doi: 10.3390/cells11111781
[40] Hernandez-Segura A, Nehme J, Demaria M. Hallmarks of Cellular Senescence[J]. Trends Cell Biol, 2018, 28(6): 436-453. doi: 10.1016/j.tcb.2018.02.001
[41] Zanotti S, Decaesteker B, Vanhauwaert S, et al. Cellular senescence in neuroblastoma[J]. Br J Cancer, 2022, 126(11): 1529-1538. doi: 10.1038/s41416-022-01755-0
[42] Van Vliet T, Varela-Eirin M, Wang B, et al. Physiological hypoxia restrains the senescence-associated secretory phenotype via AMPK-mediated mTOR suppression[J]. Mol Cell, 2021, 81(9): 2041-2052. doi: 10.1016/j.molcel.2021.03.018
[43] Bhardwaj G, Penniman CM, Klaus K, et al. Transcriptomic Regulation of Muscle Mitochondria and Calcium Signaling by Insulin/IGF-1 Receptors Depends on FoxO Transcription Factors[J]. Front Physiol, 2022, 12: 779121. doi: 10.3389/fphys.2021.779121
[44] Solyga M, Solari F. DAF-2 receptor signalling pathway (Insulin/IGF-1), a key role in muscular aging[J]. Med Sci (Paris), 2020, 36(10): 938-941. doi: 10.1051/medsci/2020166
[45] López-Otín C, Pietrocola F, Roiz-Valle D, et al. Meta-hallmarks of aging and cancer[J]. Cell Metab, 2023, 35(1): 12-35. doi: 10.1016/j.cmet.2022.11.001
[46] Liu J, Wang L, Wang Z, et al. Roles of Telomere Biology in Cell Senescence, Replicative and Chronological Ageing[J]. Cells, 2019, 8(1): 54. doi: 10.3390/cells8010054
[47] Gao B, Wang Y, Lu S. Cellular senescence affects energy metabolism, immune infiltration and immunotherapeutic response in hepatocellular carcinoma[J]. Sci Rep, 2023, 13(1): 1137. doi: 10.1038/s41598-023-28436-z
[48] Salam R, Saliou A, Bielle F, et al. Cellular senescence in malignant cells promotes tumor progression in mouse and patient Glioblastoma[J]. Nat Commun, 2023, 14(1): 441. doi: 10.1038/s41467-023-36124-9
 下载:
下载:
计量
- 文章访问数: 1695
- HTML全文浏览量: 963
- PDF下载量: 582

