
ALDH5A1 Downregulation Promotes Tumor Metastasis and Contributes to Poor Prognosis in Ovarian Cancer
-
摘要:目的
探讨醛脱氢酶5A1(ALDH5A1)在卵巢癌组织中的表达、临床意义及其影响卵巢癌细胞侵袭转移的作用和分子机制。
方法首先基于GEO数据库比较ALDH5A1在卵巢癌转移组织和原发灶的表达差异。然后通过转染小干扰RNA(siRNA)方式建立ALDH5A1表达下调的卵巢癌细胞株SKOV3,划痕实验和Transwell侵袭实验检测细胞迁移侵袭能力的变化。cBioPortal数据库描绘了ALDH5A1在卵巢癌细胞株和卵巢癌患者中的共表达基因谱。最后运用TCGA和GEO数据库分析ALDH5A1表达水平与卵巢癌预后的相关性,HPA数据库再次确认卵巢癌患者中ALDH5A1和MMPs的相对表达水平。
结果相比于卵巢癌原发灶,转移组织中的ALDH5A1表达下调。细胞实验结果显示下调ALDH5A1表达可促进卵巢癌细胞株迁移和侵袭。富集分析发现ALDH5A1的共表达基因在细胞外基质(ECM)组织通路中显著富集,进一步通过数据库转录组数据验证ECM组织通路中发挥重要作用的基质金属蛋白酶(MMP)表达与ALDH5A1表达水平呈负相关,提示ALDH5A1可能通过ECM组织通路影响卵巢癌的转移和侵袭能力。生存分析结果提示ALDH5A1低表达的卵巢癌患者预后更差。
结论ALDH5A1表达下调可能促进卵巢癌侵袭转移,且与预后不良相关。
Abstract:ObjectiveTo investigate the expression level and clinical significance of ALDH5A1 in ovarian cancer (OC) tissues, as well as to explore the possible mechanism associated with the invasion and migration of OC cells.
MethodsWe initially compared ALDH5A1 expression in metastatic tissues and the primary site of OC based on the GEO database. Then, wound-healing and Transwell assays were utilized to determine the biological role of OC cells transfected with ALDH5A1 siRNA. To unravel the potential mechanism of ALDH5A1 meditating the metastasis of OC, the coexpression profile of ALDH5A1 in OC cell lines and OC patients were generated using cBioPortal. Moreover, the TCGA and GEO databases were used to analyze the relationship between ALDH5A1 expression and the prognosis of OC patients. The HPA database was further used to confirm the relative expression of ALDH5A1 and MMPs in OC patients.
ResultsALDH5A1 expression was downregulated in metastatic tissues compared with the primary site of OC, and ALDH5A1 knockdown promoted the malignant behavior of OC cells. Additionally, the coexpression profile of ALDH5A1 was significantly enriched in the extracellular matrix (ECM) organization pathway. Western blot assay further confirmed that the expression of MMP, which played an important role in the ECM pathway, was negatively correlated with ALDH5A1 expression in OC. These results indicated that ALDH5A1 may participate in the metastasis and invasion of OC via the ECM organization pathway. Finally, KM survival plots revealed that the survival rates of OC patients with lower ALDH5A1 expression were obviously lower.
ConclusionALDH5A1 downregulation may promote the tumor metastasis and contribute to poor prognosis in OC.
-
0 引言
目前我国人口平均寿命约为76岁,在此之前的死亡称为“非成熟死亡”。降低非成熟死亡是医学工作的重中之重。原发性肝癌(primary liver cancer, PLC)居我国恶性肿瘤所致非成熟死亡原因的第一位[1-2]。我国人口占全球人口的1/5,乙肝病毒(hepatitis B virus, HBV)慢性感染占全球的1/3,PLC发病和死亡均占全球的1/2。我国大陆地区PLC中肝细胞癌(hepatocellular carcinoma, HCC)占93.0%、肝内胆管癌(intrahepatic cholangiocarcinoma, ICC)占4.3%,肝细胞癌-胆管癌混合型(combined hepatocellular-cholangiocarcinoma, CHC)占1.6%[3]。HCC只发生在有慢性损伤的肝脏中,能够导致慢性肝损伤和慢性炎性反应的疾病和行为,均可增加HCC风险。目前HCC主要危险因素有HBV慢性感染、黄曲霉素暴露、丙型肝炎病毒慢性感染、慢性脂肪性肝炎、非酒精脂肪肝、酒精性肝病、糖尿病、肝吸虫感染和马兜铃酸暴露等。HBV慢性感染是我国HCC的主要病因。HBV-HCC比其他原因引起HCC早发病10~12年,并具有更高比例的微血管侵袭(microvascular invasion, MVI)、更多子灶。HBV复制促进HCC不良预后,说明HBV除了通过引发慢性炎性反应和肝硬化以促进HCC发生外,还有直接促HCC转移的作用。其他致癌因素如黄曲霉素暴露、肝吸虫等只有在HBV慢性感染基础之上才能加快HCC发生。我国HBV慢性感染也是其他组织类型PLC的主要病因。在我国大陆地区,HCC、ICC和CHC中HBV阳性率分别是84.4%、38.6%和77.1%[3]。PLC恶性程度高,目前手术切除和肝移植是主要治疗手段。但是术后复发率高,即使是单个直径小于2 cm的HCC,术后5年复发率也达到68%,早期复发患者的预后较远期复发患者差[4]。转移是HCC预后最重要的影响因素,门静脉癌栓(portal vein tumor thrombosis, PVTT)和MVI是HCC肝内转移最主要的方式,是HCC不良预后预测的最主要指标[3-5]。细胞癌变到复发转移依赖于细胞不断的进化过程。癌细胞本身所经历的“变异-选择-适应”的进化过程依赖于肿瘤微环境(tumor microenvironment, TME)对体细胞变异的促进、选择和适应。阐明HBV致HCC的主要机制,探索有效的靶向驱动通路的新免疫-靶向联合治疗技术,对提高HCC患者生存、降低HCC非成熟死亡具有重要意义。
1 不同病因的HCC遗传特征及其与转移的关系
1.1 HBV-HCC及其他原因相关HCC基因组变异特点
和正常细胞相比,癌细胞基因组发生了系列改变,包括染色体扩增、重排和基因变异。这些改变赋予肿瘤细胞掠夺营养、无序生长特性。癌细胞的恶性程度取决于其本身特性和致癌因素。HBV不但促进HCC的发生,而且促进HCC转移和术后复发。HBV致癌机制有三要素:HBV变异赋予更高的致癌能力;HBV复制致更持久的炎性反应;HBV基因整合失活肿瘤抑制基因,激活癌症促进基因表达,尤其是C末端截断型HBx(C-terminal truncated HBx, Ct-HBx)基因整合到端粒酶逆转录酶(telomerase reverse transcriptase, TERT)、甲基转移酶(MLLs)等[3, 6-7]。不同病因导致的HCC体细胞变异谱有一定区别。中国人HCC常见体细胞单个碱基替换变异中,受炎性反应因子诱导的胞苷脱氨酶AID/APOBEC3s和老年相关的C > T/G > A转换频率最高,达23.1%。涉及基因主要有TP53(突变率达56.5%)、TERT(编码端粒酶逆转录酶,突变率45.2%)、CTNNB1(编码β-catenin,突变率22.6%)和ARID1A(突变率9.5%),TP53和TERT联合变异率最高,突变率达32.1%[8]。TP53(R249S、V157F、Y220S和P250R)和CTNNB1基因变异频率最高,SWI/SNF染色质重塑复合体重要组成亚单位(ARID1A、ARID1B和ARID2)、组蛋白甲基转移酶(MLL4和MLL3)在50%的HCC中出现变异,HBV在TERT位点发生整合率最高,CTNNB1变异与HBV关系不明显[9]。SWI/SNF染色体重塑复合体中ARID1A最常发生突变,在HCC中是常见的肿瘤抑制基因。外显子测序分析发现,ARID1A编码区变异(Q1212L和Y1211)可以在13%的HBV-HCC样本中被检出;敲低ARID1A促进HCC细胞迁移和侵袭。在HCC发生发展过程中,促进HCC进展的mTOR通过失活ARID1A-SWI/SNF复合体功能而发挥作用[10]。HBV整合不但可以使TP53基因失活,而且HBV增强子整合到癌基因附近能够大幅度提高MYC等癌基因的表达水平,增强TERT的活性,促进HCC端粒延长,加深HCC恶性程度,使HCC好发于年轻患者,且预后不良[11]。在41.7%的ICC患者中存在HBV整合,最常见的整合位点仍然是TERT启动子,常导致TERT启动子活性大幅度提高;第二个常见的HBV整合位点是FAT2基因,该整合位点常与上皮间质转化(epithelial-to-mesenchymal transition,EMT)有关;整合在DMRTA1和LINC01239之间的HBV基因组可以通过激活mTOR/4EBP/S6K信号通路促进ICC的恶性表型[12]。癌细胞进一步变异,伴随EMT和获得干性特征,促进肿瘤转移、复发和耐药。这个过程代表了癌细胞去分化和逆向发育的过程。发生EMT和获得干性特征是肿瘤转移和术后复发的关键步骤,主要是增加了肿瘤细胞迁移、对抗肿瘤药物耐药和复发的能力。癌症干细胞(cancer stem cells, CSCs)是肿瘤组织中获得干性特征的亚克隆,具有自我更新、维持肿瘤形成能力,也可以分化成多种肿瘤细胞以支持肿瘤的生长。EMT和CSC并非完全一致。间质状态是肿瘤侵袭和扩散期必需的,但不足以诱导肿瘤转移,而后期具有干性的上皮状态是保证转移的必要因素。国内学者关于CSCs研究较多,这里不赘述[13]。就HCC来说,TP53、TERT、ARID1A和MLL4突变在HCC发生和转移中起重要作用;EMT是促进HCC细胞侵袭、迁移、肿瘤干性、发生门静脉侵袭和药物抵抗的关键。
1.2 HCC体细胞变异与肿瘤抗原性的关系
HCC发生过程中的体细胞基因突变,导致氨基酸改变,形成肿瘤新抗原。肿瘤特异性抗原和相关性抗原是肿瘤免疫治疗的关键,因为肿瘤特异性T细胞免疫反应依赖于肿瘤抗原的产生和提呈。新一代测序技术发现了包括HCC在内的多种肿瘤变异谱。肿瘤突变负荷(tumor mutational burden, TMB)常被用来衡量肿瘤新抗原数量。每兆碱基超过20个体细胞变异的肿瘤被称为高TMB肿瘤,高TMB肿瘤的肿瘤免疫原性高[14]。HCC基因组每兆碱基只有5个可以改变抗原性的非同义体细胞变异,提示HCC免疫原性中等偏下,免疫治疗效果面临挑战。
1.3 基因组外DNA在HBV-HCC进化发育中的作用
癌基因往往在基因组外DNA(extrachromoso-mal DNA particles, ecDNA)中大量扩增。通过整合RNA-seq和全基因组测序数据发现ecDNA中编码的癌基因大都是在癌症中高表达的基因。这种携带癌基因的ecDNA在恶性肿瘤中常以环状形式存在。环形ecDNA比线性ecDNA拷贝数高很多。在环形ecDNA中扩增的癌基因转录水平远高于非环状基因组,显著促进癌症的恶性表型。DNA模板拷贝数并非决定基因表达的唯一决定因素,染色体结构也影响转录调控机制的可及性。应用转座酶可及性染色质测序分析(assay for transposase accessible chromatin using sequencing, ATAC-seq)等可以确定染色质可及性,定位核小体位置。ATAC-seq联合全基因组测序可确定ecDNA中癌基因[15]。目前抗HBV治疗不能根除HBV,主要原因是肝细胞核中HBV共价闭合环形DNA(covalently closed circular DNA, cccDNA)的存在。干扰素(IFN)对cccDNA有降解作用,主要是通过诱导APOBEC3s表达,但是这个过程不但慢,而且促进APOBEC3s-诱导的HBV变异。在癌旁肝组织中,HBV cccDNA水平与HBeAg和HBV DNA载量呈正相关关系,cccDNA载量与HCC不良预后呈正相关,癌旁肝组织高载量HBV cccDNA都是HCC生存的独立危险因素[16]。
1.4 染色体不稳定性在HBV-HCC进化发育中可能的作用
染色体不稳定性(chromosomal instability, CIN)是人类肿瘤的标志。CIN主要源于染色体有丝分裂过程中的错误,导致染色体结构和数量的改变。CIN与染色体非整数倍性不同:染色体非整数倍性主要指染色体数目异常,CIN是指染色体分裂错误,两者在癌症中都常见,常常同时存在。CIN与包括肝胆肿瘤在内的恶性肿瘤分期呈正相关,在复发和转移肿瘤中富集。数量和结构性CIN常形成微核,后者以双链DNA(dsDNA)形式存在于细胞质中,激活细胞的炎性反应信号。在细胞分裂的S期,微核被穿破,dsDNA暴露于细胞质,激活机体一种称为环鸟苷一磷酸-腺苷一磷酸合成酶干扰素基因刺激分子(cyclic GMP-AMP synthase-stimulator of interferon genes, cGAS-STING)抗病毒通路。在正常细胞中,急性cGAS-STING促进Ⅰ型IFN产生,促进衰老和免疫功能,产生抗病毒作用;但在癌症细胞中,细胞质dsDNA往往抑制Ⅰ型IFN信号,替代了另外的STING依赖性炎性反应信号通路——非经典型NF-κB信号。非经典型NF-κB信号激活可介导衰老相关分泌表型(senescence-associated secretory phenotype, SASP)。CIN通过促进SASP相关细胞因子的产生,招募免疫细胞到TME,促进肿瘤发生,绕过衰老,促进癌症转移和治疗抵抗;染色体非整数倍也与TME中巨噬细胞的密度显著相关,通过激活转化生长因子(TGF-β)体现了免疫抑制性表型[17]。因此,TME中炎性反应细胞有可能对dsDNA诱发的cGAS-STING有重要影响。
2 TME中免疫相关细胞及分子对HCC转移的调控作用
2022年瑞典著名肿瘤学者Douglas Hanahan在第三次发表“癌症标志性特征”时,提出将“解锁表型可塑性”、“非突变的表观遗传重编程”和“细胞老化”作为新标志加入癌症特征序列中[18]。“解锁表型可塑性”主要指“去分化或逆向分化”,“非突变的表观遗传重编程”主要指“在TME中进行表观遗传重塑”。这与“癌症进化发育学”理论体系中“逆向发育”非常相似[19]。肿瘤细胞的进化和逆向发育,离不开TME对变异细胞的选择以及变异细胞对TME的适应过程。HCC是典型的炎性反应相关恶性肿瘤。TME是一个由非特异和特异性免疫细胞组成的复杂生态系统,在HCC发生和进展中起决定性作用。
肝脏是人体最大的免疫抑制器官。肝脏巨噬细胞(Kupffer细胞)、肝星形细胞(hepatic stellate cells, HSCs)和肝窦状内皮细胞协同维护了致耐受性的微环境。Kupffer细胞不但提呈抗原,还分泌抑制性分子如IL-10、前列腺素E2和吲哚胺2-3二氧酶-1(indoleamine 2, 3- dioxygenase-1, IDO1),促进调节性T细胞(Treg)活化;肝窦状内皮细胞表达高水平PD-L1,促进TGF-β对Treg的诱导;HSC释放肝细胞生长因子(hepatocyte growth factor, HGF),促进骨髓来源的抑制细胞(myeloid-derived suppressor cell, MDSC)和Treg细胞聚集于肝脏,并通过PD-L1诱导效应T细胞凋亡。
2.1 TME中主要免疫细胞和炎性反应分子
TME主要由细胞外基质(extracellular matrix, ECM)、辅助细胞、免疫细胞、细胞因子、趋化因子和生长因子等组成[14]。TME中辅助细胞主要有肿瘤相关纤维细胞(cancer-associated fibroblast, CAF)、HSC和血管内皮细胞;免疫细胞主要有肿瘤相关巨噬细胞(tumor-associated microphage, TAM)、肿瘤相关中性粒细胞(tumor-associated neutrophil, TAN)、Treg、抑制性B细胞、MDSCs、树突细胞(dendritic cells, DCs)、细胞毒性CD8+T细胞(cytotoxic CD8+T lymphocyte, CTL)和NK细胞。这些TME内细胞群的主要功能可分为三大类:免疫抑制、免疫调节和免疫增强。TME中,CD8+T细胞、NK、DCs以及Th1细胞因子(IL-2、IFN-γ、IL-12等)参与抗肿瘤、抗病毒免疫,抑制肿瘤转移;TAN、Treg、Th17、M2 TAMs、CAFs以及Th2细胞因子促进免疫逃逸,促进HCC转移;Treg/CD8+T、Th17/Th1比例高与TME中“阴性炎性反应”水平相关并促进HCC转移[20]。TME中肿瘤细胞与免疫相关细胞通过表达共抑制受体,包括细胞毒性T细胞相关抗原4(cytotoxic T lymphocyte associated antigen 4, CTLA4)、PD-1、TIM3和淋巴细胞活化基因3(lymphocyte-activation gene 3, LAG3)、分泌相关免疫因子或通过其他未知途径,相互交流信息,共同维持了TME稳定性。HCC的TME中免疫抑制力量要超过抗肿瘤免疫力量,整体有免疫抑制特性。
2.2 TME中主要发挥免疫抑制的细胞种类和作用机制
HCC转移主要以PVTT形式进行。在PVTT发生过程中,TME中重要调控辅助细胞-CAFs通过下调细胞外基质信号通路—核心蛋白多糖-β1整合素信号通路,促进HCC的血管侵袭。β1整合素促进HCC侵袭,而核心蛋白多糖通过降低β1整合素抑制HCC体外侵袭和迁移[21]。肿瘤来源的溶血磷脂酸可激活CAFs,CAFs产生的HGF可以调节HCC干细胞的可塑性,促进TME中的中性粒细胞和巨噬细胞等的集聚,激活如MDSCs和Treg等免疫抑制细胞,促进HCC组织中的缺氧和纤维化。CAFs产生的内皮唾液酸蛋白诱导巨噬细胞的极化,促进HCC进展。CAFs通过IL-6/STAT3信号通路激活PD-L1+ TANs和受损T细胞,促进肿瘤免疫逃逸[22]。
TAMs在HCC生长中起关键作用。巨噬细胞是HCC组织中密度最高的免疫细胞,其中M1型巨噬细胞通过刺激淋巴细胞发挥抗肿瘤作用;M2型巨噬细胞通过抑制淋巴细胞功能、诱导新血管生成,发挥促癌作用。IFN-γ和脂多糖(lipopolysaccharide, LPS)促进单核细胞向M1型巨噬细胞极化;HCC产生的细胞因子IL-4、IL-13、克隆刺激因子(colony stimulating factor, CSF-1)、CCL2、CXCL12和结缔组织生长因子(connective tissue growth factor, CTGF)促进单核细胞向M2型分化。在炎性反应性TME中,M2-TAMs促进HCC发生、侵袭和转移[14, 20]。HCC产生的骨桥蛋白促进TAMs趋化活性;HCC产生的缺氧诱导因子1α(hypoxia-inducible factor 1α, HIF-1α)促进TAMs释放IL-1β,IL-1β反向诱导HCC发生EMT。CD68+巨噬细胞在肿瘤组织中的数量显著高于癌旁肝组织,而且TAMs多数为IL-10阳性,IL-10促进肿瘤组织中CD4+CD25+Foxp3+Treg的数量,后者抑制CD8+T细胞和NK细胞对肿瘤细胞的毒性。IL-6主要由巨噬细胞等细胞产生,促使巨噬细胞向M2型转换,变成TAMs,通过活化STAT3信号通路促进HCC发生。TAM增加Th17数量,后者抑制免疫检查点治疗产生的抗肿瘤免疫。
中性粒细胞大体上可分为N1型和N2型。促进炎性反应的N1型具有抗肿瘤作用,抵抗炎性反应的N2型具有促肿瘤作用。这种差异性取决于中性粒细胞的极化方式:LPS、IFN-γ和Ⅰ类IFNs促进向N1方向分化,IL-4促进向N2方向分化。TANs在HCC生长中也起关键作用。缺氧诱导HIF-1α表达异常活跃,导致HIF-1α依赖性趋化因子上调,促进TANs在肿瘤组织中的浸润。TANs浸润和中性粒/淋巴细胞百分比代表了全身性“阴性”炎性反应,促进肿瘤进展及HCC不良预后[20]。N2-TANs对抗肿瘤免疫的特别抑制作用主要是通过抑制CD8+T细胞功能并诱导CD8+T细胞凋亡。
MDSCs是分布于TME中不成熟的骨髓细胞,表现出强烈的免疫抑制特性。MDSCs主要通过精氨酸酶、可诱导性一氧化氮合成酶(inducible nitric oxide synthase, iNOS)、致免疫耐受性的酶如IDO1、活性氧簇(reactive oxygen species, ROS)、TGF-β和IL-10抑制CTLs和NK活性。MDSC主要有两个亚群:单核细胞的MDSC(M-MDSC)和多形核的MDSC(PMN-MDSC)。在TME中,M-MDSC比PMN-MDSC具有更高的免疫抑制活性。一些来自癌细胞的细胞因子G-CSF、GM-CSF、VEGF、MCP-1和IL-1促进MDSCs在HCC组织中浸润。局部慢性缺氧可以通过趋化因子CCL26/CX3CR1通路诱导MDSCs集聚于HCC组织中,损伤CD8+效应T细胞的功能,降低NK细胞毒性和细胞因子分泌,诱导Treg,显著抑制免疫并对HCC进展起关键促进作用。外周血中MDSCs预示HCC的预后不良[14]。在HCC细胞转移定殖到目标组织前,肿瘤细胞还分泌某些可溶性因子包括CXCL17、G-CSF、骨桥蛋白、CXCL12、TNF-α、TGF-β、VEGF-A和PIGF。这些因子通过影响CD11b+/Gr-1+MDSC介导的新血管生成、诱导炎性反应、重塑ECM以及募集免疫细胞,营造免疫抑制但营养丰富的TME。这些抑制性免疫细胞密度高,促进HCC复发和转移,与HCC不良预后有关,符合“冷肿瘤”特点,免疫治疗难以奏效。
2.3 TME中抗肿瘤免疫细胞的种类和作用机制
肿瘤相关免疫细胞的功能取决于免疫诱导的方向。CD4+T辅助细胞(Th)的分化取决于炎性反应细胞因子的诱导,既可以分化成Th1,促进CD8+T细胞形成和成熟,参与抗病毒、抗肿瘤免疫;也可以分化成Treg和Th17,具有免疫抑制作用,促进肿瘤转移。这种差异取决于Th1细胞因子(IL-12、IL-2、IFN-γ)和Th2细胞因子(IL-4、IL-10、IL-13、TGFβ、IL-17)的平衡。Th1细胞因子促进Th0向Th1分化;Th2细胞因子促进Th0向Th2、Treg和Th17分化。Treg虽然在免疫稳态中起关键作用,但是在肿瘤免疫中主要起抑制作用。
HCC组织中的树突细胞(dendritic cells, DCs)是主要的肿瘤抗原提呈细胞,主要分DC-c1-CD1C、DC-c3-CLEC9A和DC-c4-LAMP3三类,DC-c1-CD1C高表达CD1C、FCER1A和CLEC10A,相当于常规DC2(cDC2);DC-c3-CLEC9A高表达CLEC9A、XCR1和CADM1相当于常规DC1(cDC1);DC-c4-LAMP3表达成熟标志物LAMP3、CD80和CD83、迁移标志物CCR7和淋巴细胞再循环趋化因子CCL19和CCL21。血中幼稚型cDC1、cDC2和类浆细胞DCs(pDCs)并不表达LAMP3,但是体外经过LPS联合IFN-γ或poly I: C刺激后成熟,表达LAMP3。LAMP3+DCs在肿瘤组织中表达高于癌旁肝组织,具有高度吞噬活性,在肿瘤引流淋巴结中通过受体和配体结合方式刺激CD8+T、CD4+T细胞和NK细胞的免疫反应[23]。CD8+T细胞和NK细胞具有直接细胞毒活性和明确的抗肿瘤作用,在TME中密度高则与HCC预后良好密切相关。CD8+T细胞和NK细胞在肿瘤组织中的存在代表了“热肿瘤”特征,免疫治疗会有抗癌效果。
3 TME免疫细胞的代谢重编程对HCC转移的潜在作用
3.1 TME中缺氧通过代谢重编程影响免疫平衡
线粒体功能是CD8+T细胞扩增、产生细胞因子和有效免疫记忆不可或缺的。NKT细胞发挥功能严重依赖线粒体的氧化磷酸化。因此,抗肿瘤免疫需要在有氧环境中发挥作用。相反,在HCC这种高糖酵解的肿瘤中,Treg通过单羧酸盐转运因子1积极利用糖酵解产生的乳酸,促进活化T细胞核因子1转位进入细胞核内,增强Treg的PD-1表达,进一步加强免疫抑制[24-25]。肿瘤迅速生长与氧供应渠道缺乏导致缺氧在TME普遍存在。缺氧导致乳酸增加造成的酸性环境促进Treg富集、分化和发挥功能,起到免疫抑制作用。TME富含乳酸,而且乳酸可提升肿瘤浸润性Treg的复制能力和免疫抑制功能,导致免疫抑制;葡萄糖对TME中Treg的功能和扩增的作用与乳酸相反[26]。因此,调节TME中Treg能量代谢有助于了解能量代谢在HCC转移中的作用,有利于发现控制HCC转移的新途径。
3.2 TME中缺氧通过ECM重编程对HCC进展的影响
缺氧环境可降低甲基胞嘧啶过氧化酶的活性,导致某些超甲基化的产生;慢性缺氧可导致HIF-1α表达,促进TME新血管生成,缺氧还可以通过上调EMT主要调控分子ZEB1以诱导组蛋白甲基转移酶SETD1B表达,后者又以正反馈方式维持ZEB1表达,促进HCC细胞进入侵袭性EMT状态;TME中缺氧和基质细胞炎性反应因子还可以通过上调EMT核心调控分子SNAIL1表达,引起染色质重塑,诱导EMT、提升HCC细胞的侵袭能力[14]。缺氧能诱导一些分子表达,如赖氨酰氧化酶(lysyl oxidase, LOX),能促进胶原蛋白和弹性蛋白的共价交联和纤连蛋白(fibronectin, FN1)的表达,促进HCC复发转移,预测不良预后[27]。FN1不但在ECM和血清中高表达促进HCC转移,也可以在有血管侵袭的Ⅵ期转移性或晚期HCC细胞中高表达。其表达水平不但可以诊断HCC,且具有良好的敏感度和特异性(在cut off值为100 µg/ml时,FN1诊断HCC的敏感度达92%,特异性达88%),还可以准确预测HCC不良预后。不过FN1和HCC转移的相关性与HCC病因无关[28]。适度的持续性缺氧对HCC的进化发育有一定促进作用。
3.3 犬尿氨酸代谢通路对TME免疫抑制的影响
色氨酸是人体只能从食物中获取的8种必需氨基酸之一,在细胞生长和功能维持中起重要作用。90%的色氨酸是通过犬尿氨酸通路(kynurenine pathway, KP)代谢,生成调节细胞代谢过程的关键辅酶:尼克酰胺腺嘌呤二核苷酸(NAD+)。在正常条件下,KP受到严格调控,但在炎性反应TME中,KP通路高度激活,迅速降解色氨酸。色氨酸是Th和CD8+CTL生存和扩增的必需氨基酸。色氨酸缺乏导致CTL扩增受阻,免疫前体细胞向Treg方向分化,导致TME免疫抑制。KP通路始于3个限速酶:IDO1、IDO2和色氨酸2, 3-二氧酶(tryptophan 2, 3-dioxygenase, TDO2),能催化色氨酸转化为犬尿氨酸,后者在犬尿氨酸3加单氧酶(kynurenine 3-monooxygenase, KMO)等一系列酶作用下最终生成NAD+。在生理条件下,IDO1的表达局限于肺和胎盘的内皮细胞;IDO2主要在肝细胞、胆管细胞、神经细胞和肾脏中表达;TDO2主要在肝脏中表达。犬尿氨酸是KP通路的第一个代谢物,作为内源性配体,以自分泌和旁分泌形式激活芳香烃受体(aryl hydrocarbon receptor, AhR)。AhR是配体激活的PAS家族基础螺旋-环-螺旋的转录因子,在多种免疫细胞中表达,发挥重要免疫调节作用。在恶性肿瘤发生发展过程中,炎性反应因子如IL-6可以促进肿瘤细胞表达IDO1,IDO1可通过激活炎性反应因子正反馈环“IDO-AhR-IL-6-STAT3”信号通路促进肿瘤的生长和转移。在IDO1/IDO2高表达的肿瘤细胞中,TME中的色氨酸被剥夺,免疫监督功能被强烈抑制。IDO2在胶质母细胞瘤、乳腺癌、结直肠癌和肝癌中表达,显著促进免疫逃逸、促进癌细胞扩增和转移。激活AhR促进癌细胞扩增、组织浸润和转移。犬尿氨酸-AhR信号通路抑制免疫细胞的分化和活性,促进肿瘤免疫耐受。色氨酸的二级代谢物犬尿喹啉酸也可以作为AHR内源性配体,在IL-1β存在的情况下诱导IL-6的产生,促进炎性反应正反馈环的形成。KP通路的代谢产物包括犬尿氨酸本身均可抑制T细胞扩增和活化,扰乱Th和Treg之间的平衡,使其向Treg方向发展,导致CTL耗竭,促进肿瘤免疫逃逸。TME中某些“毒性”代谢产物,包括乳酸、犬尿氨酸和腺苷等在塑造免疫抑制特性和肿瘤免疫逃逸方面起重要作用。但是,KP通路在HCC研究中有相互矛盾之处:IDO1只在IFNγ刺激的HCC中高表达,HCC中IDO1高表达与CD8+T细胞密度呈正相关;而TDO2在HCC中高表达与HCC生长、侵袭和转移密切相关[29]。AhR的高表达与HCC扩增和侵袭有关,促进EMT进程,参与HCC的进化发育。
4 HBV在致癌过程中与免疫抑制性TME的关系
前文提到,与HCV慢性感染等其他原因相关的HCC相比,HBV-HCC早发病10~12年,而且预后更差[3],说明HBV除了导致炎性反应之外,还有直接促HCC作用。HBV致癌主要依赖HBV进化、整合和复制(包括cccDNA载量)。我们研究发现HBV变异是在炎性反应基础上,IL-6反式激活促变异分子APOBEC3B的表达、抑制变异修复分子UNG的表达,使APOBEC3B-UNG之间的平衡向促突变方向倾斜,导致HBV变异和体细胞变异增加[30]。APOBEC3B-UNG之间的平衡在胆囊癌、胆管癌和肾细胞癌进化发育中均起重要作用,但是组织特异性反式因子和炎性反应因子在调控该平衡中起重要调控作用[31-32]。HBV变异在癌组织、癌旁肝组织和外周血中进化不同步,在癌组织中因为免疫功能最弱,进化最慢,在外周血中进化最强,免疫选择的变异倾向于促进癌症发生。HBV进化不但在促进HCC中发挥重要作用,也显著促进HCC的不良预后[6, 33]。变异型HBV致癌能力增强,主要是通过增强促癌炎性反应所致,IL-5和IL-6在变异型HBx基因转入“睡美人转座子”动物模型中表达水平显著高于野生型;而且HBx基因变异型主要通过激活血浆酶原激活物抑制因子1(plasminogen activator inhibitor-1,PAI1)和细胞分裂周期20(CDC20)促进HCC发生和进展[34]。PAI1,又称SERPINE1,能诱导肿瘤新血管生成,促进肿瘤细胞扩散和转移,是多种恶性肿瘤重要治疗靶标。PAI1在肿瘤组织中高表达是HCC恶性程度和不良预后的主要标志。CAFs诱导M2-TAMs并促进HCC的作用是通过PAI1促进HCC的EMT而实现[35]。我们还发现,HBV前S区变异通过激活STAT3/IL-6传统炎性反应通路,促进HCC发生和转移。因此,HBV进化可通过PAI1和STAT3/IL-6等,以“正反馈”方式放大炎性反应促癌的效能,以改造HCC的TME,促进HCC的进化发育。HBV整合是随机的,但是整合到能促进HCC发生发展的位点才有机会在HCC中被发现。HBV整合促进HCC的位点主要是灭活TP53基因、激活TERT启动子、激活MLL4甲基化转移酶,促进HCC进化发育。
5 癌症进化发育学在PLC特异性预防和靶向联合免疫治疗中的应用
HBV致癌的研究揭示了“癌症进化发育学”基本规律:HLA-II类抗原遗传多态性决定了CD4+T细胞免疫走向,促进了HBV慢性炎性反应化,“非可控性炎性反应”因子如IL-6反式作用导致促进变异的APOBEC3s与变异修复的UNG之间平衡的失调,使平衡向APOBEC3s方向倾斜;AID/APOBEC3s胞苷脱氨酶家族,尤其是APOBEC3B促进体细胞和病毒变异;由于遗传和环境因素导致CD8+T细胞清除HBV能力较弱,选择了促癌HBV变异。携带关键体细胞变异的肝细胞经过“炎性反应-坏死-增生”在受损肝脏中积累突变、促进染色体重塑和表观遗传改变、转化为初始HCC细胞。非可控性炎性反应参与决定了HBV变异的免疫选择方向;作为HBV复制模板,HBV cccDNA应携带HBV变异,后者在细胞质中形成dsDNA或与人基因组DNA片段组成环ecDNA,在肝细胞中激活cGAS-STING信号,诱导Ⅰ型IFN抗病毒作用;在癌变后,dsDNA促进慢性cGAS-STING下游非经典NF-κB激活,产生TGF-β1、PAI1、Th2等细胞因子,召集和武装MDSC、TAM、Treg和中性粒细胞等免疫抑制性炎性反应细胞,形成促进HCC进化发育的TME;而这些抑制性免疫细胞通过分泌IL-10、TGF-β1和产生ROS等抑制CD8+T细胞、NK细胞和DC等抗病毒、抗肿瘤免疫活性,还通过分泌IL-1β和IL-6等促进肝癌细胞逆向分化;与相应野生型HBV相比,促癌HBV变异在体内能诱导更加严重的促癌炎性反应,放大了“炎-癌”转化效应。HBV-HCC进展离不开病毒整合变异,两者协同促进变异细胞逆向发育。HCC起始细胞在炎性反应TME中获得生长优势,造成缺氧,诱导HIF-1α和VEGF等表达,促进新血管生成。同时癌细胞改变了自身代谢模式,以糖酵解方式为主要供能模式的同时,产生戊糖直接用于核酸合成;缺氧和乳酸堆积促进Treg扩增、抑制NK和CTL,营造免疫抑制TME。这些抑制性免疫细胞进一步促进早期HCC发生EMT并向干细胞方向进化,促进转移和术后复发[19]。虽然我们是基于HBV致癌研究提出了“癌症进化发育学”基本理论框架,但该理论中的主要步骤如APOBEC3B-UNG平衡、免疫平衡等在几乎所有肿瘤类型中均存在,反映了癌症发生和转移的基本规律[36]。
6 免疫抑制性TME与HCC免疫检查点治疗和靶向治疗的相互作用
6.1 免疫抑制性TME对HCC免疫检查点治疗和靶向治疗的影响
HCC的TME基本是免疫抑制性的,主要包括负向免疫调控的Treg、抑制性B细胞、MDSCs、M2-TAMs等免疫抑制细胞,共刺激淋巴细胞信号包括免疫检查点配体和受体,IDO或精氨酸酶等致耐受性的酶类[14, 37]。这些免疫抑制因素与促进肿瘤免疫的CTL、NK细胞和DCs之间的平衡决定了肿瘤免疫治疗的效果,诱导有效抗肿瘤免疫必须突破免疫抑制屏障。免疫检测点包括表达在效应淋巴细胞上的共抑制分子,包括CTLA4、PD-1、TIM3和LAG3等[38]。CTLA4(又称CD152)表达在激活T细胞,尤其是Treg表面,和CD28共享配体,其激活预防自身免疫。PD-1(又称CD279)表达在激活的受损T细胞的细胞膜,与表达在基质细胞、肿瘤细胞和某些髓样细胞表面的配体PD-L1特异性结合,特异性抑制效应T细胞功能,导致效应T细胞耗竭或失能。免疫检查点抑制剂(immune checkpoint inhibitors, ICIs)是能够阻断这些受体与其配体结合的单克隆抗体,阻止T细胞失活。
目前免疫检测点阻断治疗在晚期HCC治疗中有一定的客观疗效。单独使用对晚期HCC有7%~20%的总有效率,中位生存时间12.9~15.1月[14]。HCC免疫抑制性TME和抗原识别受损的肿瘤-免疫相互作用可促进肿瘤免疫逃逸。在TME中,Treg和CTL之间的平衡以及Treg和NK细胞之间的平衡是肿瘤进展和肿瘤免疫的关键。T细胞的数量和活化状态无疑对肿瘤进展和ICIs的疗效有重要影响。β-catenin变异的HCC往往淋巴细胞浸润较少,ICIs治疗难以奏效。肿瘤细胞产生的某些分子如TGF-β1、IL-10、IDO、AFP等通过刺激MDSCs和Treg细胞,促进TME的免疫抑制功能,显著限制了ICIs的疗效。M2-TAM直接或间接通过抑制细胞毒性免疫细胞如CD8+T细胞和NK细胞,促进HCC进展,显著限制了免疫治疗的疗效。对抗HCC中TGF-β1、IDO或VEGF的免疫抑制活性有助于提高ICIs疗效。
6.2 靶向治疗对TME免疫抑制性的影响
靶向治疗一线药物是在Sorafenib的基础上,应用一种靶向血管内皮细胞生长因子受体VEGFR1-3、纤维母细胞生长因子受体FGFR1-4以及PDGFR2的小分子多激酶抑制剂Lenvatinib,可以使晚期HCC的中位生存时间达到13.6个月。动物模型研究发现,Lenvatinib单药治疗可显著提升NK细胞比例,降低CD4+T细胞比例;Lenvatinib和EGFR抑制剂Gefitinb联合使用可显著增加NK细胞和CD8+T进入肿瘤体内的数量,降低TAMs的比例[39]。可见靶向治疗可增强免疫治疗效果,两种治疗策略有一定的互补作用。目前最有效的HCC治疗方式是免疫与靶向联合治疗,如晚期HCC最有效的一线治疗是应用单抗atezolizumab阻断PD-1联合单抗bevacizumab阻断VEGF,整体有效率可达36%,中位生存时间达17个月[14]。虽然靶向和免疫治疗在晚期HCC中取得了令人瞩目的疗效,但是大部分患者并没有生存获益,而且生存获益患者也存在快速复发和转移问题。因此,需要进一步阐明TME中免疫微环境对HCC转移和复发的作用机制,丰富和发展“癌症进化发育学”理论体系,从而使HCC这种恶性程度很高的肿瘤得以变成可控制的慢性病,达到大幅度降低HCC非成熟死亡率。
综上,我们基于HBV致癌研究提出的“癌症进化发育学”理论体系,不但揭示了炎性反应或病毒致癌的部分重要机制,为晚期肝癌的靶向和免疫治疗奠定了理论基础、提供了技术方法,同时有希望为其他恶性肿瘤的防治提供参考。
Competing interests: The authors declare that they have no competing interests.作者贡献:赵桐:研究设计、实验实施和论文撰写田训:研究思路指导和论文审较曹晨:研究设计、实验指导和论文审校 -
![]()
图 1 ALDH5A1下调促进卵巢癌细胞侵袭与转移
Figure 1 ALDH5A1 downregulation promoted metastasis and invasion of ovarian cancer
![]()
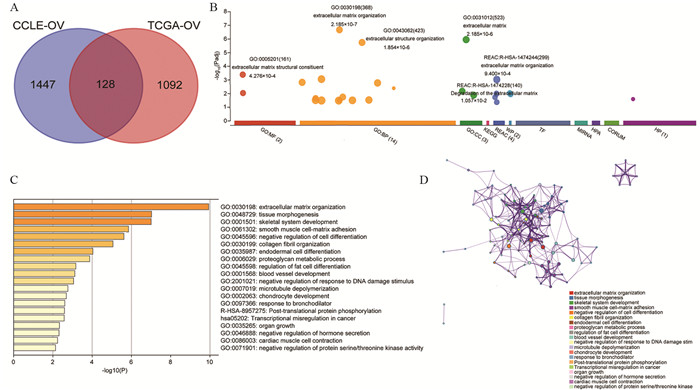
图 2 ALDH5A1基因功能富集分析以及其与卵巢癌中的共表达基因
Figure 2 Gene-Ontology analysis of ALDH5A1 and coexpressed genes revealing the relationship between ALDH5A1 and ECM signaling pathways in OC
![]()
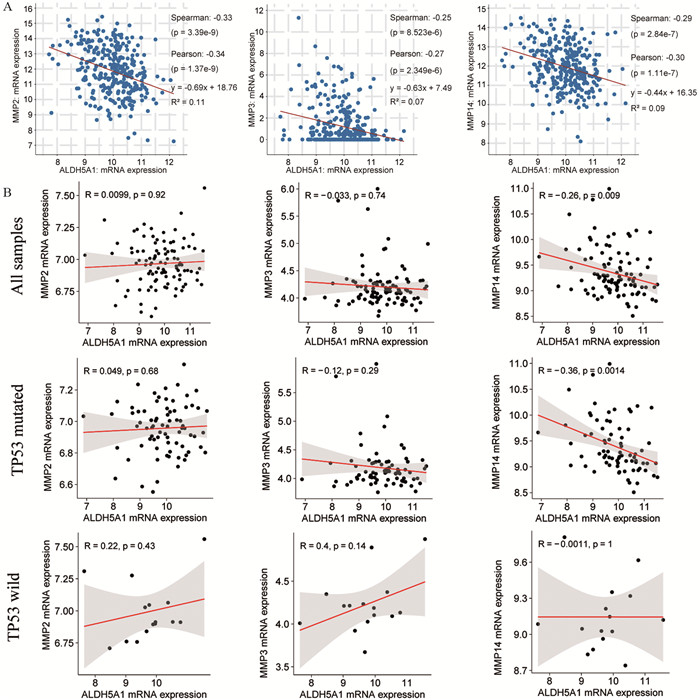
图 3 ALDH5A1与ECM组织通路的共表达分析
Figure 3 Coexpression analysis of ALDH5A1 and correlated genes among ECM pathways
![]()
图 4 HPA数据库中两例代表性卵巢癌患者免疫组织化学提示ALDH5A1和MMP14表达负相关
Figure 4 Immunohistochemical staining results from HPA database demonstrating the negative correlation between ALDH5A1 and MMP14
-
[1] Sung H, Ferlay J, Siegel RL, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries[J]. CA Cancer J Clin, 2021, 71(3): 209-249. doi: 10.3322/caac.21660
[2] Torre LA, Trabert B, Desantis CE, et al. Ovarian cancer statistics[J]. CA Cancer J Clin, 2018, 68(4): 284-296. doi: 10.3322/caac.21456
[3] Yang Y, Karakhanova S, Hartwig W, et al. Mitochondria and Mitochondrial ROS in Cancer: Novel Targets for Anticancer Therapy[J]. J Cell Physiol, 2016, 231(12): 2570-2581. doi: 10.1002/jcp.25349
[4] Zhou Z, Ibekwe E, Chornenkyy Y. Metabolic Alterations in Cancer Cells and the Emerging Role of Oncometabolites as Drivers of Neoplastic Change[J]. Antioxidants (Basel), 2018, 7(1): 16. doi: 10.3390/antiox7010016
[5] Egan G, Khan DH, Lee JB, et al. Mitochondrial and Metabolic Pathways Regulate Nuclear Gene Expression to Control Differentiation, Stem Cell Function, and Immune Response in Leukemia[J]. Cancer Discov, 2021, 11(5): 1052-1066. doi: 10.1158/2159-8290.CD-20-1227
[6] Hilvo M, De Santiago I, Gopalacharyulu P, et al. Accumulated Metabolites of Hydroxybutyric Acid Serve as Diagnostic and Prognostic Biomarkers of Ovarian High-Grade Serous Carcinomas[J]. Cancer Res, 2016, 76(4): 796-804. doi: 10.1158/0008-5472.CAN-15-2298
[7] Deng XY, Gan XX, Feng JH, et al. ALDH5A1 acts as a tumour promoter and has a prognostic impact in papillary thyroid carcinoma[J]. Cell Biochem Funct, 2021, 39(2): 317-325. doi: 10.1002/cbf.3584
[8] Kaur H, Mao S, Li Q, et al. RNA-Seq of human breast ductal carcinoma in situ models reveals aldehyde dehydrogenase isoform 5A1 as a novel potential target[J]. PLoS One, 2012, 7(12): e50249. doi: 10.1371/journal.pone.0050249
[9] Tian X, Han Y, Yu L, et al. Decreased expression of ALDH5A1 predicts prognosis in patients with ovarian cancer[J]. Cancer Biol Ther, 2017, 18(4): 245-251. doi: 10.1080/15384047.2017.1295175
[10] Goswami CP, Nakshatri H. PROGgeneV2: enhancements on the existing database[J]. BMC Cancer, 2014, 14: 970. doi: 10.1186/1471-2407-14-970
[11] Cerami E, Gao J, Dogrusoz U, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data[J]. Cancer Discov, 2012, 2(5): 401-404. doi: 10.1158/2159-8290.CD-12-0095
[12] Raudvere U, Kolberg L, Kuzmin I, et al. g: Profiler: a web server for functional enrichment analysis and conversions of gene lists (2019 update)[J]. Nucleic Acids Res, 2019, 47(W1): W191-W198. doi: 10.1093/nar/gkz369
[13] Zhou Y, Zhou B, Pache L, et al. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets[J]. Nat Commun, 2019, 10(1): 1523. doi: 10.1038/s41467-019-09234-6
[14] Nicholson-Guthrie CS, Guthrie GD, Sutton GP, et al. Urine GABA levels in ovarian cancer patients: elevated GABA in malignancy[J]. Cancer Lett, 2001, 162(1): 27-30. doi: 10.1016/S0304-3835(00)00620-0
[15] 胡梅艳, 孙晓红. 细胞外基质、基质金属蛋白酶与恶性肿瘤关系的研究进展[J]. 肿瘤药学, 2016, 6(1): 26-30. https://www.cnki.com.cn/Article/CJFDTOTAL-LIYX201601008.htm Hu YM, Sun XH. The research progress of relationship between ECM, MMPs and malignant tumor[J]. Zhong Liu Yao Xue, 2016, 6(1): 26-30. https://www.cnki.com.cn/Article/CJFDTOTAL-LIYX201601008.htm
[16] Wang X, Yang B, She Y, et al. The lncRNA TP73-AS1 promotes ovarian cancer cell proliferation and metastasis via modulation of MMP2 and MMP9[J]. J Cell Biochem, 2018, 119(9): 7790-7799. doi: 10.1002/jcb.27158
[17] Gonzalez-Villasana V, Fuentes-Mattei E, Ivan C, et al. Rac1/Pak1/p38/MMP-2 Axis Regulates Angiogenesis in Ovarian Cancer[J]. Clin Cancer Res, 2015, 21(9): 2127-2137. doi: 10.1158/1078-0432.CCR-14-2279
[18] Qiu J, Ye L, Ding J, et al. Effects of oestrogen on long noncoding RNA expression in oestrogen receptor alpha-positive ovarian cancer cells[J]. J Steroid Biochem Mol Biol, 2014, 141: 60-70. doi: 10.1016/j.jsbmb.2013.12.017
[19] Castro-Castro A, Marchesin V, Monteiro P, et al. Cellular and Molecular Mechanisms of MT1-MMP-Dependent Cancer Cell Invasion[J]. Annu Rev Cell Dev Biol, 2016, 32: 555-576. doi: 10.1146/annurev-cellbio-111315-125227
[20] Kaimal R, Aljumaily R, Tressel SL, et al. Selective blockade of matrix metalloprotease-14 with a monoclonal antibody abrogates invasion, angiogenesis, and tumor growth in ovarian cancer[J]. Cancer Res, 2013, 73(8): 2457-2467. doi: 10.1158/0008-5472.CAN-12-1426
 下载:
下载:



计量
- 文章访问数: 1963
- HTML全文浏览量: 726
- PDF下载量: 287

