 2016, Vol. 43
2016, Vol. 43文章信息
- 陈颖,甘露,张幸平.
- CHEN Ying, GAN Lu, ZHANG Xingping.
- 外周型原始神经外胚叶瘤临床特征及治疗分析
- Clinical Characteristics and Treatment Methods on Peripheral Primitive Neuroectodermal Tumors Patients
- 肿瘤防治研究, 2016, 43(01): 25-29
- Cancer Research on Prevention and Treatment, 2016, 43(01): 25-29
- http://www.zlfzyj.com/CN/10.3971/j.issn.1000-8578.2016.01.006
-
文章历史
- 收稿日期: 2015-08-18
- 修回日期: 2015-10-04
引用本文 |



原始神经外胚叶瘤(primitive neuroectodermal tumors,PNET)于1973年由Hart首次提出,认为是一种罕见的由神经嵴衍生的具有多向分化潜能的小圆细胞恶性肿瘤[1]。按肿瘤生长部位不同,PENT分为中央型原始神经外胚叶瘤(central primitive neuroectodermal tumors,cPNET)和外周型原始神经外胚叶瘤(peripheral primitive neuroectodermal tumors,pPNET)[2]。pPNET发病率低、恶性度高,Kelleher等[3]报道美国1973~2004年间pPNET年发病率为2.93/100万。查阅文献,国内外多以个案报道或结合文献的病例分析为主。本研究对重庆医科大学附属第一医院收治的10例pPNET患者资料进行总结分析,对其临床病理特征及治疗方法进行探讨,旨在为pPNET的临床诊治提供参考。
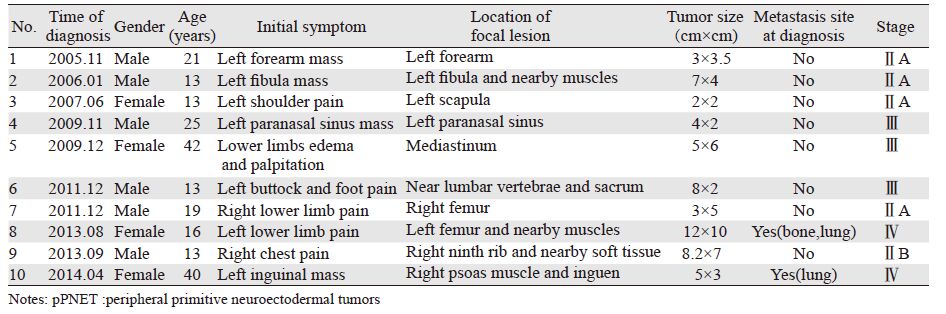
1 资料与方法 1.1 临床资料回顾性分析重庆医科大学附属第一医院2005年11月—2014年4月收治的10例经病理组织学、免疫组织化学确诊为pPNET患者的临床、病理、治疗及随访资料。纳入标准:(1)经手术或活检病理组织学、免疫组织化学确诊为PNET;(2)按照肿瘤生长部位,诊断为pPNET;(3)有完整的临床、病理、治疗及随访资料。排除标准:(1)结合临床资料,诊断为cPNET;(2)确诊后未在本院接受治疗的患者;(3)合并其他肿瘤性疾病者。按照美国癌症联合委员会(AJCC)第7版的肿瘤分期标准进行分期:ⅡA期4例,ⅡB期1例,Ⅲ期3例,Ⅳ期2例,见表 1。
1.2 研究方法采用手工检索重庆医科大学附属第一医院2005年11月—2014年4月住院病历记录。以“pPNET”或“外周型原始神经外胚叶瘤”为检索词,共检索出10例符合纳入标准的病例。提取患者基本信息、临床特征、影像学及血液学检查结果、病理特征以及治疗经过等资料,运用电话随访、门诊复查相结合的方式进行随访,随访截至2015年5月,随访率为100%。重点观察患者治疗后有无肿瘤复发、复发时间、患者生存时间、死亡原因,采用直接计算法计算患者1、2、5年生存率及平均生存时间。
2 结果 2.1 临床基本特征本组病例中男6例,女4例。年龄13~42岁,平均年龄21.5岁。患者主要症状为局部包块、疼痛,1例肿瘤位于纵隔者以双下肢肿胀、心悸为首发症状。肿瘤累及部位:鼻窦1例、躯干4例、四肢5例。肿瘤大小:最小为2 cm×2 cm,最大为12 cm×10 cm。初诊时1例伴肺转移,1例伴肺、骨转移,见表 1。
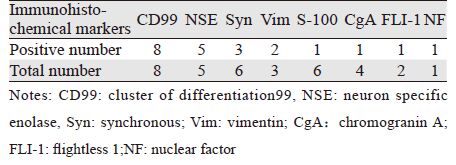
病理学特征:肉眼观可见实性肿块,与周围组织粘连,常伴有邻近骨质破坏,部分有包膜,部分伴有出血、坏死或黄白色黏稠液体。光学显微镜下肿瘤细胞多呈大小一致的小圆细胞,瘤细胞呈片状或巢状分布,胞体小、胞质少、有液泡,核深染、呈圆形或椭圆形,见图 1。免疫组织化学结果,见表 2、图 2。5例检测Ki67表达率分别为10%、30%、35%、45%及70%。

|
| The tumor cells of pPNET were composed of uniform small round cells A: HE ×200; B: ×400 图 1 外周型原始神经外胚叶瘤组织HE染色结果 Figure 1 HE results of pPNET tissues |

|
| A: CD99(+); B:NSE(+); C: Syn(+); D: FLI-1(+) 图 2 外周型原始神经外胚叶瘤细胞免疫组织化学CD99、NSE、Syn、FLI-1的表达情况 (SP ×400) Figure 2 Immunohistochemical expression of CD99, NSE, Syn and FLI-1 in pPNET cells (SP ×400) |
本组病例行CT、MRI、PET/CT或B型超声等影像学检查,均提示局部软组织肿块影、密度或信号不均匀,与邻近组织分界不清,部分伴有周围骨质破坏,增强CT或MRI呈不均匀强化,见图 3。初诊时50%(5/10)患者血清乳酸脱氢酶(LDH)升高至正常值的2~3倍;50%(5/10)患者外周血白细胞(WBC)数升高,以中性粒细胞升高为主,最高者白细胞为11.8×109/L,中性粒细胞高达91%。随着肿瘤手术切除或放疗、化疗后,LDH、WBC等指标均有所下降。

|
| A: CT 3D reconstrucion; B: T1WI; C: T2WI; D: T2WI-STIRA: CT scan showed a mass in the marrow cavity of right femur with bone destruction nearby; B-D: MRI showed the mass in the left leg had the similar T1 signal on the T1W1. T2W1 showed mainly long T2 signal. T2WI-STIR showed higher signal 图 3 外周型原始神经外胚叶瘤患者影像学检查结果 Figure 3 Imaging t results of pPNET patients |
10例患者中5例行手术+术后放疗+化疗,其中1例治疗10月后出现原发部位肿瘤复发,另4例无瘤生存至今。2例行根治性放疗+化疗,其中1例治疗18月后出现肿瘤复发再行化疗。1例行单纯手术治疗,治疗3月后出现肿瘤复发行再次手术及化疗。1例行化疗+高强度聚焦超声(high intensity focused ultrasound,HIFU)治疗,1例行中药治疗。在7例行放疗的患者中,2例行根治性放疗、5例行术后放疗。7例均采用局部扩大野、常规分割放疗,根治性放疗剂量:62 Gy/31 f,术后放疗剂量:58 Gy/29 f~60 Gy/30 f。化疗方案多以长春新碱、环磷酰胺/异环磷酰胺、表柔比星、放线菌素D、依托泊苷、达卡巴嗪等为基础的2~4药联合化疗6~8周期,见表 3。
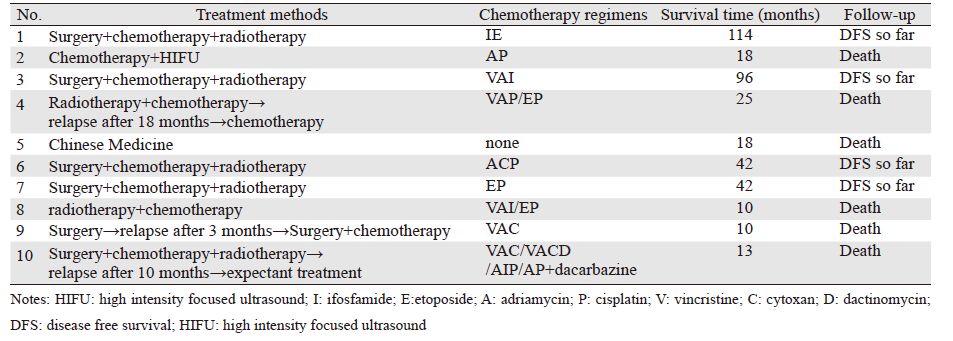
至随访截止时间,10例中有6例死亡(均死于肿瘤),4例无瘤生存至今。本组患者初诊时伴远处转移2例(见表 1和表 3中的例8、例10),分别在治疗后10月、13月死亡。治疗后局部肿瘤复发3例,其中1例为手术+放化疗、1例为单纯手术者、1例为放化疗后复发(见表 3中的例10、例9、例4),平均复发时间为首次治疗后10.3月,3例均在挽救性治疗后死亡。另2例死亡患者中1例行化疗+HIFU治疗,1例仅行中药治疗(见表 3中的例2、例5)。6例死亡患者的生存时间为10月~25月,平均生存时间15.7月,死亡原因为肿瘤局部复发或进展(3例为肿瘤复发,3例为肿瘤进展)。4例至今无瘤生存中,生存时间为42月~114月,平均生存时间73.5月。全组平均生存时间38.8月(范围10月~114月)。1年生存率为80.0%(8/10),2年生存率为71.4%(5/7),5年生存率为40.0%(2/5)。
3 讨论pPNET是一种临床少见恶性肿瘤,以青少年男性居多,好发部位以躯干中轴、四肢及周围软组织多见,少数可发生于实质脏器[4]。在本组病例中,患者平均年龄为21.5岁,男性占60%(6/10),肿瘤多位于四肢及躯干中轴,与文献[4]报道相似。pPNET的临床表现、影像学检查缺乏特异性,因肿瘤原发部位、浸润生长方式不同,临床表现各异。本组大部分病例以局部包块伴疼痛就诊,也有因肿瘤原发部位特殊,表现出相应的临床症状,如1例因肿瘤位于纵隔近心脏后壁区域以心悸、双下肢肿胀为首发症状。文献报道,pPNET患者初诊时20%~30%伴有远处转移,常见转移部位有肺、骨和骨髓[5]。本组10例患者中初诊时有2例伴有骨转移和(或)肺转移,与文献[5]报道相符。在本组6例死亡的晚期患者中,3例血清LDH值比正常高2~3倍。朱金莲等[6]认为LDH增高是pP NET的不良预后因素,对肿瘤复发转移有重要的监测意义。
pPNET光学显微镜下瘤细胞通常为大小一致的小圆细胞,典型者呈Homer-Wright假玫瑰花团样排列,部分非典型者为多形性大细胞型,以梭形细胞多见[7]。目前细胞形态学联合免疫组织化学是诊断pPNET的主要方法。其中CD99曾作为公认的敏感指标,但后来研究发现其特异性不高;而FLI-1敏感度为70%~80%,特异性为90%,故临床上常用CD99结合FLI-1或两种以上神经分化标志物(如NSE、Syn、S-100、NF)等来诊断pPNET[8, 9, 10]。本组病例免疫组织化学中CD99、NSE阳性表达高(分别为8/8、5/5),Syn、S-100阳性表达分别为3/6、1/6,NF检测1例阳性。因FLI-1检测近几年才开展,在本组患者中FLI-1检测2例,1例为阳性。EWS/pPNET患者中超过85%有t(11; 22)(q24; q12)染色体易位,形成EWS/FLI-1融合基因[11]。检测EWS/FLI-1融合基因及其特异性染色体易位的敏感度达91%,特异性达100%,已逐渐成为诊断EWS/pPNET的金标准[12]。肿瘤的分子病理诊断依赖于特异性、敏感度较高的分子标志物,但检测费用高、方法较复杂,故该融合基因检测尚未广泛应用于临床。在我院10例患者中,均未行EWS/FLI-1融合基因检测,需要今后进一步改进和完善。
pPNET的治疗多参考尤文氏肉瘤治疗原则。2015年版尤文氏肉瘤NCCN指南推荐对于局灶EWS/pPNET病变的治疗模式:至少12周新辅助化疗+局部治疗(手术+放疗)+28~49周维持辅助化疗+辅助放疗。有文献报道,pPNET经综合治疗后5年生存率约60%(52%~88%)[13]。
对于早期局限性pPNET患者,能够达到广泛切除或切缘阴性时,应首选手术治疗。李瑞健等[5]报道33例pPNET患者行根治术、姑息性手术及未手术的平均中位生存时间分别为30月、8月及6月。进一步行单因素(P=0.001)及多因素(P=0.017)分析显示,手术方式(根治术、姑息性手术和未手术)对总生存率的影响有显著差异。本组有6例行手术治疗,生存时间在10月~114月,平均生存时间52.8月。有4例未行手术治疗(1例位于鼻窦,1例位于纵隔、靠近主动脉等大血管,1例因初诊时就出现骨、肺等脏器转移,故不适宜手术;1例因要求保肢,不愿行手术治疗),生存时间在10月~25月,平均生存时间17.8月。行手术治疗者平均生存时间较未行手术者延长35月,明显提高总生存率,与文献报道相符。但pPNET多呈侵袭性生长,患者就诊时多属晚期,往往手术不能完全切除,肿瘤局部复发率高,需进行术后辅助治疗。
放射治疗适用于手术不能切除、切除不彻底或切缘阳性的患者,旨在降低肿瘤局部复发率。根治性放疗剂量55~60 Gy,术后放疗剂量45~55 Gy[14]。对于肿瘤完全切除者术后放疗是否获益,欧洲与北美学者并未达成一致。目前研究推荐:pPNET肉眼可见肿瘤剂量55.8 Gy,显微镜残留病灶剂量50.4 Gy,每次1.8~2 Gy[14]。本组有7例行放疗(2例根治性放疗、5例术后放疗),均采用局部扩大野常规分割放疗,其中根治性放疗剂量62 Gy/31 f,术后放疗剂量58 Gy/29 f~60 Gy/30 f。随访至今,根治性放疗2例均出现肿瘤局部复发;术后放疗5例中,1例在治疗10月后出现肿瘤复发,另4例均无瘤生存至今 。 Biswas等[15]报道pPNET对放疗较敏感,但单独放疗并不能成功治愈pPNET患者。结合文献报道及我院病例,我们认为放疗联合手术综合治疗pPNET的肿瘤局部控制率及患者预后较单纯手术或单纯放疗好。本组放疗剂量与文献报道比较剂量偏高,但仍出现肿瘤复发,故pPNET的最佳放疗剂量还需进一步探讨。
化疗可以缩小pPNET肿瘤病灶、提高患者保肢率及生存率。2015年版尤文氏肉瘤NCCN指南推荐,pPNET的化疗以异环磷酰胺(I)/环磷酰胺(C)、依托泊苷(E)、阿霉素(A)/放线菌素D及长春新碱(V)的2~4药联合化疗方案,化疗维持时间总共在28~49周。临床试验证实VACD方案的局部控制率及5年无病生存率(DFS)均优于VAC方案(局控率为96% vs. 86%,5年DFS为60% vs. 24%),而VACD/IE交替治疗方案可获得更高的5年DFS(69%)[16]。在本组9例化疗病例中,均以常规剂量辅助化疗为主。化疗方案有VAC、VAI、IE、VAP、ACP、EP等方案。本组病例中,由于化疗不良反应较重,总疗程数均未达到指南所推荐的数目,可能是导致5年生存率较文献报道低的原因。
分子靶向治疗是近年来肿瘤学领域的研究热点。Prochilo等[17]报道VEGFR-2通路阻断会使pPNET肿瘤细胞生长及血管生成受抑制,而EWS/FLI-1被认为是VEGF启动子,运用舒尼替尼治疗表达此融合基因的pPNET患者,增加了放射敏感度。复习近8年国内外文献,关于pPNET的靶向治疗目前尚处于临床研究阶段,报道不多。作为pPNET中表达率达85%左右的EWS/FLI-1融合基因可能为该病的分子靶向治疗提供一个方向。
影响pPNET患者预后的因素有:青少年起病、肿瘤位于四肢、小于2 cm、无远处转移、LDH值正常及Ki67阳性率低者提示预后相对较好[6, 15]。在治疗上,早期诊断、完全手术切除、多学科综合治疗者预后较好,其中完全手术切除是影响患者预后的独立因素[18]。本组病例中,目前仍无瘤生存的4例均为青少年起病(13~21岁),分期较早(ⅡA期3例,Ⅲ期1例),均行手术切除并接受过术后放疗、化疗等综合治疗。本组预后不良者多在2年内死亡,生存时间分别为10月2例、13月1例、18月2例、25月1例,平均生存时间为15.7月。6例死亡患者中2例分期较早(ⅡA、ⅡB期各1例,见表 1和表 3中的例2、例9),估计与患者未接受综合治疗有关。而预后较好的患者中,平均生存时间为73.5月,最长1例至今已无瘤生存114月。
综上所述,外周型原始神经外胚叶瘤发病率低、恶性度高,国内外文献报道较少。pPNET患者以青少年男性为主,临床及影像学表现缺乏特异性,以病理组织学联合免疫组织化学确诊,CD99、NSE、Syn表达率高,FLI-1及EWS/FLI-1融合基因检测将提高其诊断的特异性及敏感度。pPNET的治疗以手术、放疗、化疗综合治疗为主,分子靶向治疗有待深入探索。pPNET患者的预后根据初诊时临床病理特征、临床分期及治疗情况等因素共同决定。
| [1] | Cole M, Parajuli S, Laske D, et al. Peripheral primitive neuroectodermal tumor of the dura in a 51-year-old woman following intensive treatment for breast cancer[J]. Am J Case Rep, 2014, 15: 294-9. |
| [2] | Zhang Y, Li H. Primitive Neuroectodermal Tumors of Adrenal Gland[J]. Jpn J Clin Oncol, 2010, 40(8): 800-4. |
| [3] | Kelleher FC, Thomas DM. Molecular pathogenesis and targeted therapeutics in Ewing sarcoma/primitive neuroectodermal tumours[J]. Clin Sarcoma Res, 2012, 2(1): 6. |
| [4] | Dutta D, Shivaprasad KS, Das RN, et al. Primitive neuroectodermal tumor of adrenal:clinical presentation and outcomes[J]. J Cancer Res Ther, 2013, 9(4): 709-11. |
| [5] | Li RJ, Zhao LJ, Wang P, et al. Peripheral primitive neuroectodermal tumor: clinical analysis of 33 cases[J]. Zhongguo Zhong Liu Lin Chuang, 2011, 38(15): 910-4. [李瑞健, 赵路军, 王平, 等. 33例外周原始神经外胚瘤临床分析[J]. 中国肿瘤临床, 2011, 38(15): 910-4.] |
| [6] | Zhu JL, Yuan SX, Wang XL, et al. Analysis of primitive neuroectodermal tumor’s prognostic factors[J]. Zhonghua Zhong Liu Fang Zhi Za Zhi, 2014, 21 (3): 228-31. [朱金莲, 袁苏徐, 王晓丽, 等. 原始神经外胚层瘤预后相关因素分析[J]. 中华肿瘤防治杂志, 2014, 21(3): 228-31.] |
| [7] | Desai SS, Jambhekar NA. Pathology of Ewing′s sarcoma/PNET: Current opinion and emerging concepts[J]. Indian J Orthop, 2010, 44(4): 363-8. |
| [8] | Tsokos M, Alaggio RD, Dehner LP, et al. Ewing sarcoma/peripheral primitive neuroectodermal tumor and related tumors[J]. Pediatr Dev Pathol, 2012, 15(1 Suppl): 108-26. |
| [9] | Hafezi S, Seethala RR, Stelow EB, et al. Ewing’s Family of Tumors of the Sinonasal Tract and Maxillary Bone[J]. Head Neck Pathol, 2011, 5(1): 8-16. |
| [10] | Li Q, Cui W, Abulajiang G, et al. Application of immunohistochemistry in the diagnosis of small round blue-cell tumors of soft tissue[J]. Clin Lab, 2014, 60(8): 1383-92. |
| [11] | Potratz J, Jürgens H, Craft A, et al. Ewing sarcoma: biology-based therapeutic perspectives[J]. Pediatr Hematol Oncol, 2012, 29(1): 12-27. |
| [12] | Shingde MV, Buckland M, Busam KJ, et al. Primary cutaneous Ewing sarcoma/primitive neuroectodermal tumour: a clinicopathological analysis of seven cases highlighting diagnostic pitfalls and the role of FISH testing in diagnosis[J]. J Clin Pathol, 2009, 62(10): 915-9. |
| [13] | Natale R, Thariat J, Vedrine PO, et al. Conservative multimodal management of a primitive neuroectodermal tumor of the thyroid[J]. Rare Tumors, 2013, 5(2): 75-8. |
| [14] | Bernstein M, Kovar H, Paulussen M, et al. Ewing’s sarcoma family of tumors:current management[J]. Oncologist, 2006,11(5): 503-19. |
| [15] | Biswas B, Rastogi S, Khan SA, et al. Developing a prognostic model for localized Ewing sarcoma family of tumors: A single institutional experience of 224 cases treated with uniform chemotherapy protocol[J]. J Surg Oncol, 2015, 111(6): 683-9. |
| [16] | Grier H. Addition of ifosfamide and etoposide to standard chemotherapy for Ewing’s sarcoma and primitive neuroectodermal tumor of bone[J]. N Engl J Med, 2003, 348(8): 694-701. |
| [17] | Prochilo T, Savelli G, Bertocchi P, et al. Targeting VEGF-VEGFR Pathway by Sunitinib in Peripheral Primitive Neuroectodermal Tumor, Paraganglioma and Epithelioid Hemangioendothelioma: Three Case Reports[J]. Case Rep Oncol, 2013, 6(1): 90-7. |
| [18] | Zhang FC, Tang L, Ma Y, et al. Analysis of clinical features and prognosis in 126 patients with peripheral primitive neuroectodermal tumor[J]. Shanghai Jiao Tong Da Xue Xue Bao(Yi Xue Ban), 2012, 32(11): 1490-6. [张凤春, 唐雷, 马越, 等. 126例外周性原始神经外胚层瘤临床特征及预后因素分析[J]. 上海交通大学学报(医学版), 2012, 32(11): 1490-6.] |