 2015, Vol. 42
2015, Vol. 42文章信息
- 刘雪咏,张声,陈余朋,王行富,王鹏程,李国平,李晓玲.
- LIU Xueyong, ZHANG Sheng, CHEN Yupeng, WANG Xingfu, WANG Pengcheng, LI Guoping, LI Xiaoling.
- 散发型脑膜血管瘤病7例临床病理分析
- Sporadic Meningioangiomatosis: A Clinicopathological Analysis of Seven Cases
- 肿瘤防治研究, 2015, 42(11): 1131-1134
- Cancer Res Prev Treat, 2015, 42(11): 1131-1134
- http://www.zlfzyj.com/CN/10.3971/j.issn.1000-8578.2015.11.016
-
文章历史
- 收稿日期: 2015-01-20
- 修回日期: 2015-04-23
引用本文 |



2. 364000 龙岩,福建省龙岩市第二医院病理科
Department of Pathology, The Second Hospital of Longyan, Longyan 364000, China
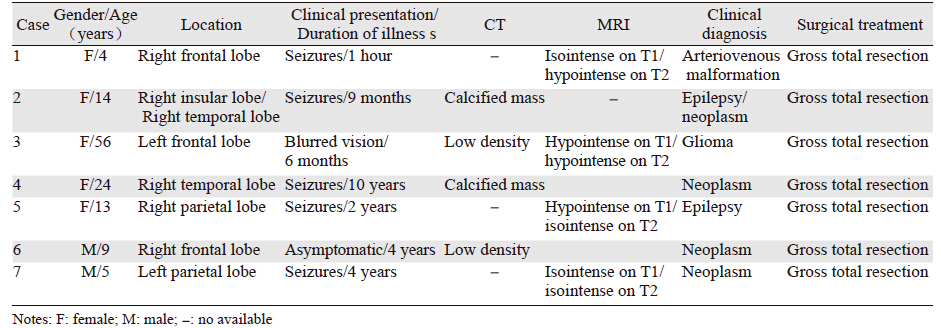
回顾性分析福建医科大学附属第一医院2012年6月—2014年11月手术治疗的7例MA。7例MA均为散发型,其中5例发生于右侧大脑半球,2例发生于左侧大脑半球。男性2例,女性5例,男女之比2:5。年龄4~56岁,平均17.86岁。首发症状为癫痫发作者5例,视物模糊者1例,发现肿块但无症状者1例,病程从1小时~10年。7例病例中收集到4例CT和4例MRI检查资料,其中6例表现为占位性病变。CT检查2例显示高密度钙化影,1例显示稍低密度影,1例显示低密度影;MRI检查结果分别为等T1短T2、长T1短T2、长T1等T2、等T1等T2各1例;对比增强扫描3例未见明显强化,1例明显均匀强化。典型病例例3见图 1。术前临床诊断困难,无一例诊断明确。7例均行肿瘤全切术,术后均未行放化疗。随访时间1~29月,均无复发,截止本次随访,所有病例无瘤生存,见表 1。

|
| MRI showed a lesion in the left frontal lobe (case 3), which was hypointensity on both T1(A)- and T2-(B) weighted imaging without enhancement(C) 图 1 散发型脑膜血管瘤病患者的MRI Figure 1 MRI f inding s o f spo r adi c meningioangiomatosis patients |
 |
所有标本经10%的中性缓冲福尔马林固定,石蜡包埋,3 μm厚切片行HE和免疫组织化学染色,免疫组织化学采用EnVision法染色。一抗GFAP、vimentin、EMA、PR、CD34、NeuN、Syn、p53和MIB-1等购自Dako公司。
2 结果 2.1 病理特征 2.1.1 肉眼观察结果4例送检组织为大块状脑组织,其中2例脑表面脑沟变浅,病变累及脑表面,另外2例脑表面未见明显异常,切面灰质白质交界尚清,局灶皮质呈灰白色,质韧;3例送检组织为不规则组织,切面灰白,质韧。
2.1.2 镜检结果7例病例显示了类似的组织病理学表现。病变主体均位于大脑皮质,未累及白质。3例局灶累及其上软脑膜。4例以血管增生为主,见图 2A,3例以梭形细胞增生为主,见图 2B。血管及血管周围梭形细胞与脑实质组织边界不规则,呈漩涡状、斑片状或分枝状浸润周围脑组织。血管大小较一致,分布均匀,无充血、出血征象,伴不同程度的玻璃样变。梭形细胞胞质嗜伊红,核呈圆形到伸长形,染色质较细腻。无明显细胞异型性及核分裂。皮质内充满大小不等、形态不一的钙化小体,大多数以血管为中心形成,呈同心圆状钙化为主,其他钙化模式也可见,包括点状、小片状或粉尘样钙化等。病变间区可见胶质反应性增生和残存的变性神经元伴神经原纤维缠结形成,并可见散在的Rosenthal纤维及嗜酸性颗粒小体。所有病例均无坏死。例2伴发局灶皮质发育不良,例6伴发过渡型脑膜瘤(WHOⅠ级)及周围骨质膨出,例7伴发纤维型脑膜瘤(WHOⅠ级),其余无明显伴随病变。

|
| A: histopathologically, meningioangiomatosis was composed of two main elements, proliferative small blood vessels and perivascular spindal cells (HE ×100); B: these proliferative perivascular spindle cells exhibited some histological features, similar to meningothelial cells. The nuclei were round to elongated with bland chromatin. The cytoplasm appeared eosinophilic. There was no obvious atypia or mitoses (HE ×400); C: these spindle cells were focal positive for EMA (EnVision ×400); D: GFAP-positive for reactive gliosis (EnVision ×400); E: NeuN-positive for degenerative neurons (EnVision ×400); F: Tau-positive for neurofibrillary tangles within cytoplasm of neuron (EnVision ×600) 图 2 散发型脑膜血管瘤病患者组织病理学结果 Figure 2 Histopathological findings of sporadic meningioangiomatosis patients |
血管及血管周围梭形细胞vimentin阳性;7例病例中,3例梭形细胞成分EMA局灶阳性,见图 2C,4例阴性;3例梭形细胞成分PR个别细胞阳性,4例阴性;神经胶质GFAP阳性,见图 2D;神经元Syn、NeuN阳性,见图 2E;神经元纤维缠结Tau蛋白阳性,见图 2F;病灶内细胞增殖指数Ki67低于1%。
3 讨论1915年,Bassoe和Nuzum首次描述该病变。1937年,Worster-Drought等把该病变命名为MA。随后,MA被认为是儿童和成人难治性癫痫的罕见病因之一。导致儿童顽固性癫痫的脑组织病变中,MA占2.3%[2]。部分学者认为MA无性别偏向[3],部分学者认为MA好发于男性[4],而本组病例中男女之比2:5。好发部位是右侧大脑半球,最常见于额叶和颞叶,其他罕见部位包括第三脑室、大脑脚、胼胝体、三叉神经节、脑干、小脑[5]等。MA通常散发,但大概25%的病例与NF2相关。散发型MA诊断的中位年龄是17岁;NF2相关型MA诊断的中位年龄是25岁[1]。散发型常为孤立性病变,伴有难治性癫痫发作、头痛或其他神经症状;NF2相关型常为多灶性并且无症状,一般于头部外伤后或尸检中发现。
MA最常伴发WHOⅠ级脑膜瘤,偶可伴发WHOⅡ级脑膜瘤[6]。脑膜瘤伴发MA者有类似单纯性MA的NF2/4.1B基因缺失、merlin/protein 4.1B蛋白缺失,这些结果提示脑膜瘤伴发的MA可能是一种组织学类似于MA的病变,有人认为可能是脑膜瘤沿Virchow-Robin间隙蔓延所致,但这组病变预后良好,不同于脑膜瘤浸润脑实质的生物学行为[1]。MA还可伴发其他病变,包括血管畸形、脑膨出、脑软化、局灶皮质发育不良、少突胶质细胞瘤、血管外皮细胞瘤等。MA的发病机制尚不清楚。目前存在3种假说:一是脑组织退行性改变形成的错构瘤性病变;二是皮质血管畸形诱导Virchow-Robin间隙蛛网膜内皮及纤维细胞增生所致;三是脑膜瘤脑实质侵袭所致。NF2相关型MA与NF2基因突变相关。散发型MA的遗传学分析显示几乎所有病例未见NF2基因突变。出现症状的病例一般是散发型MA。
影像学特征对MA的诊断和鉴别诊断无明显特异性。本组病例中,CT和MRI错误提示为血管畸形、胶质瘤、钙化结节等。CT扫描显示病变通常为高或者稍高信号,伴钙化,但低或正常信号亦有报道。MRI表现各异,较常表现为T1低或等信号,T2显示信号不均匀,常为低信号。病变中钙化的高密度区显示T1和T2低信号。可表现为不均匀对比增强。MA在MRI上表现脑回状高信号是重要的影像学特征[7]。
术中发现,MA不累及硬膜组织。活检组织通常质偏硬,甚至呈橡胶样,难以切开。病变在皮质表面或皮质内斑片状生长,并向周围脑组织带状扩展,一般不累及白质。微钙化导致大体呈灰白色,伴沙粒感。通常呈实性,偶见多囊性病变[8]。
典型的MA具有特征性组织病理学表现,由小血管及血管周围梭形细胞组成。有学者根据上述两种成分含量的多少,将病变分为细胞为主型和血管为主型,组织形态的不同主要取决于病变时间长短[9]。MA特征性表现是皮质浅层小血管异常增生并分支向皮质深层延伸。一般不累及皮质下白质,偶可累及软脑膜。这些增生的血管显示不同程度的玻璃样变和钙化,钙化模式多样,可表现为同心圆状、点状、片状和粉尘状等。血管周围梭形细胞增殖是MA的另一重要组织学特征。这些梭形细胞显示了一些脑膜上皮细胞的组织学特征,呈巢状或旋涡状,偶伴砂粒体形成。细胞核呈圆形到伸长形,胞质嗜伊红。核内假包涵体并不是MA的特征。皮质中这些增生的血管和梭形细胞呈岛屿状,与周围的神经组织边界不规则,呈地图状。病变中陷入的神经胶质组织反应性增生并微钙化。常可发现陷入的神经元发生退行性变,包括神经原纤维缠结、颗粒空泡变性、Pick小体出现。散发型MA与NF2相关型MA的组织学表现相似,虽然NF2相关型MA更可能出现血管广泛玻璃样变,及多量透明变组织取代大部分脑皮质。
MA组织起源尚不明确。因其组织学特征可表现为巢团状、漩涡状生长模式及上皮样细胞形态,偶伴砂粒体形成及EMA阳性,目前普遍认为MA起源于脑膜上皮细胞。本组7例病例中3例表现为EMA局灶阳性,提示可能起源于脑膜上皮。但由于仅25%的病例表达EMA, 并且常是弱或者局灶表达,且梭形细胞不均匀表达CK,偶可表达S-100[10],这与脑膜上皮免疫组织化学特征并不完全一致。有学者通过超微结构观察,结果也不能明确血管周围梭形细胞来源于脑膜上皮细胞[4]。另外,部分学者认为MA可能是脑膜上皮细胞、血管和成纤维细胞错构性的增殖[11];部分学者认为是血管畸形伴脑膜上皮细胞反应性增生[12];部分学者认为是多潜能细胞分化而来[9]。
MA需与如下病变相鉴别:(1)脑膜瘤浸润脑组织:临床症状进展较快,影像学表现为明显的占位效应和瘤周水肿,增强扫描呈明显均匀强化。邻近脑膜增厚并强化,呈鼠尾状改变,即“脑膜尾征”。镜下常表现为肿瘤细胞不规则舌状突入、浸润脑组织,破坏皮质结构,肿瘤细胞增殖指数较高。大部分脑膜瘤表达EMA。(2)血管畸形:血管大小不等、厚薄不一,常伴充血、出血,管壁可玻璃样变性、钙化,未见血管周围梭形细胞增殖。(3)婴儿促纤维增生型星形细胞瘤和节细胞胶质瘤:好发于1岁以下的儿童。它们具有特征性的影像学表现,常表现为大囊小结节,并囊壁强化。这些病变没有像MA那样具有明显的血管增生。(4)Sturge-Weber综合征:是以眼部、皮肤及脑血管瘤为主要表现的先天性遗传性疾病。与MA最具鉴别诊断的特征是Sturge-Weber综合征常伴皮质萎缩。
完全切除术预后良好,术后一般不复发,癫痫症状得以控制。新近文献报道,完全切除术后癫痫症状治愈率可达80%,仅20%术后需辅助抗癫痫药物治疗[13]。
| [1] | Perry A, Kurtkaya-Yapicier O, Scheithauer BW, et al. Insights into meningioangiomatosis with and without meningioma: a clinicopathologic and genetic series of 24 cases with review of the literature[J]. Brain Pathol, 2005, 15(1): 55-65. |
| [2] | Prayson RA. Tumours arising in the setting of paediatric chronic epilepsy[J]. Pathology, 2010, 42(5): 426-31. |
| [3] | Tacconi L, Thom M, Symon L. Cerebral meningioangiomatosis: case report[J]. Surg Neurol, 1997, 48(3): 255-60. |
| [4] | Wiebe S, Munoz DG, Smith S, et al. Meningioangiomatosis. A comprehensive analysis of clinical and laboratory features[J]. Brain, 1999, 122 ( Pt 4): 709-26. |
| [5] | Omeis I, Hillard VH, Braun A, et al. Meningioangiomatosis associated with neurofibromatosis: report of 2 cases in a single family and review of the literature[J]. Surg Neuro, l2006, 65(6): 595-603. |
| [6] | Chen YY, Tiang XY, Li Z, et al. Sporadic meningioangiomatosis-associated atypical meningioma mimicking parenchymal invasion of brain: a case report and review of the literature[J]. Diagn Pathol, 2010, 5: 39. |
| [7] | Yao Z, Wang Y, Zee C, et al. Computed tomography and magnetic resonance appearance of sporadic meningioangiomatosis correlated with pathological findings[J]. J Comput Assist Tomogr, 2009, 33(5): 799-804. |
| [8] | Li P, Cui G, Wang Y, et al. Multicystic meningioangiomatosis[J]. BMC Neurol, 2014, 14: 32. |
| [9] | Koutsopoulos AV, Yannopoulos A, Stathopoulos EN, et al. Meningioangiomatosis with predominantly cellular pattern[J]. Neuropathology, 2003, 23(2): 141-5. |
| [10] | Yasha TC, Ghosal N, Singh SS, et al. Meningioangiomatosis: a report on a rare case, masquerading as schwannoma[J]. Indian J Pathol Microbiol, 2012, 55(1): 117-8. |
| [11] | Louis DN, Stemmer-Rachamimov AO, Wiestler OD. Neurofibromatosis type 2[M]//P Kleihues, WK Cavenee. World Health Organization Classification of Tumours: Pathology and Genetics: Tumours of the Nervous System[M]. Lyon: IARC Press, 2000: 219-22. |
| [12] | Blumenthal D, Berho M, Bloomfield S, et al. Childhood meningioma associated with meningioangiomatosis:case report[J]. J Neurosurg, 1993, 78(2): 287-9. |
| [13] | Feng R, Hu J, Che X, et al. Diagnosis and surgical treatment of sporadic meningioangiomatosis[J]. Clin Neurol Neurosurg, 2013, 115(8): 1407-14. |